

patofiziologiya_sist_krovi_2010
.pdfМеханизм окклюзионного криза связан с гипоксической или стрессорной провокацией агрегации ненормальных эритроцитов и соответствующими микроциркуляторными нарушениями в различных органах (легкие, селезенка, почки, мозг). При секвестрационном кризе развивается внезапная спленомегалия вследствие переполнения органа кровью.
Кривая Прайс-Джонса сдвигается вправо вследствие повышенного образования молодых эритроцитов.
По этиологии различают первичные (наследственные) и вторичные (приобретенные) гемолитические анемии.
Наследственные гемолитические анемии
Наследственные гемолитические анемии относятся к эн-
доэритроцитарным ГА и согласно классификации Стокмена (1992) возникают в результате наследования:
1)дефектов структуры мембран эритроцитов (мембранопатии, или эритроцитопатии);
2)патологических типов гемоглобинов (гемоглобинопатии);
3)дефицита ферментов (ферментопатии, или энзимопатии).
К мембранопатиям относятся гемолитические анемии, обусловленные генетическим дефектом белковой или липидной структуры мембраны, цитоскелета эритроцитов. Клеточная мембрана эритроцита состоит из двойного липидного слоя, на ее поверхности эксперессированы антигены. Цитоскелет эритроцитов состоит из спектрина, актина, белка полос 4.1, 4,2 и анкирина (рис. 23). Цитоскелет обеспечивает эритроциту двояковогнутую форму и необходимую деформируемость, что позволяет ему проходить через отверстия диаметром 2,5-3 мкм.
Наследственные дефекты спектрина и ассоциированных с ним белков (например, анкирина) сопровождаются увеличением риска нарушения целостности мембраны в условиях выраженной гипоксии, сопряженной с дефицитом АТФ и увеличением содержания катионов кальция в клетках.
91
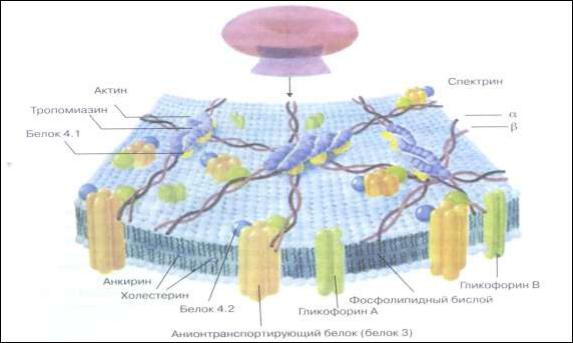
Рис. 23. Схематическое изображение внутренней стороны мембраны эритроцита с сетью миофиламентных белков, формирующих цитоскелет
К белковозависимым мембранопатиям относят наследственный микросфероцитоз (болезнь Минковского-Шоффара), эллиптоцитоз (овалоцитоз), стоматоцитоз; к липидозависимым мембранопатиям – наследственный акантоцитоз.
- Болезнь Минковского-Шоффара (микросфероцитоз) –
вторая по распространенности среди всех наследственных ГА наследуется аутосомно-доминантно и встречается только среди представителей белой расы. Мембранопатия обусловлена снижением содержания и изменением структуры примембранных белков эритроцитов (спектрина и анкирина), нарушением взаимодействия их с другими белками мембраны эритроцита, что приводит к повышению ее проницаемости, гипергидратации клеток. Реже болезнь вызвана дефектами других подмембранных белков цитоскелета – протеина 4.2 и белка третьей полосы. Эритроциты приобретают сферическую форму, при этом уменьшается их диаметр (до 5,4 мкм) – развивается микросфероцитоз (рис.24), уменьшается осмотическая стойкость.
Снижение деформируемости микросфероцитов приводит к их внутриклеточному гемолизу (в синусоидах селезенки) развивается спленомегалия. Продолжительность жизни микросферо-
92
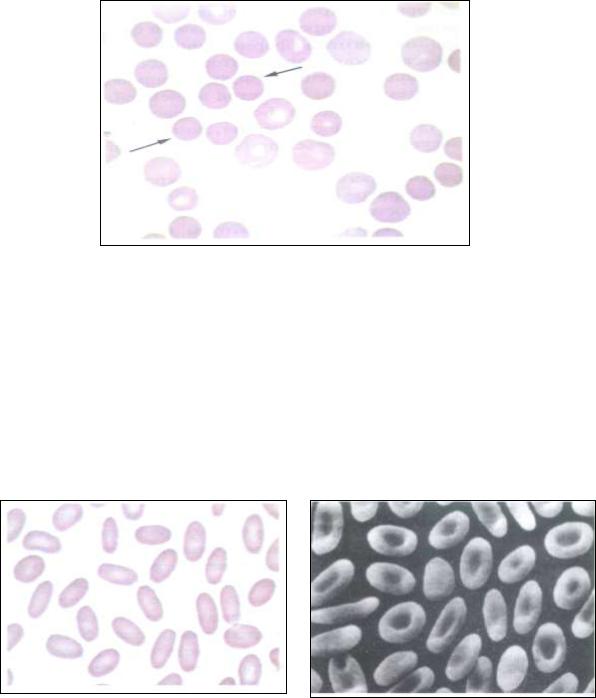
цитов составляет 8-15 суток. Это заболевание является одной из важнейших причин врожденной гемолитической желтухи.
Рис. 24. Микросфероцитоз. Болезнь Минковского-Шоффара
- Эллиптоцитоз (овалоцитоз) также наследуется аутосом- но-доминантно. Мембранопатия обусловлена дефектом мембранного белка 4.1. Образуются эритроциты овальной или элиптической формы (до 25-75 % от общего количества эритроцитов) (рис.25, 26).
Рис. 25. Овалоцитоз. |
Рис. 26. Овалоцитоз. |
Периферическая кровь |
Сканирующая электронная |
|
микроскопия |
93
- Стоматоцитоз наследуется аутосомно-доминантно. В основе заболевания лежит нарушение структуры мембраны. Эритроциты в центре содержат неокрашенный участок в виде светлой полосы, напоминающей рот (от греч. stoma – рот) (рис.27).
Рис.27. Стоматоцитоз
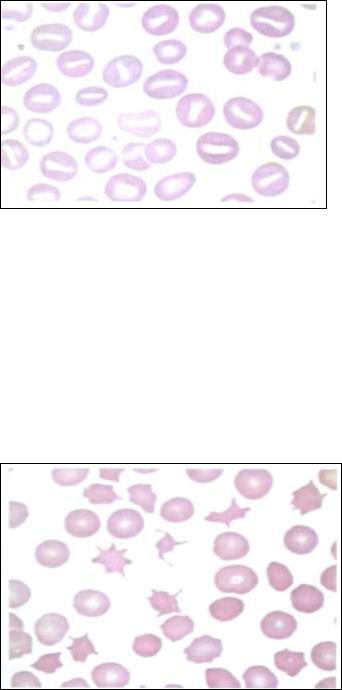
- Акантоцитоз наследуется аутосомно-рецессивно. Заболевание связано с нарушением липидного обмена. Снижение содержания триглицеридов, фосфолипидов в крови отражается на липидном составе мембран. Нарушения в эритроцитах способствуют снижению текучести мембран и изменению их формы. Эритроциты приобретают выпячивания мембраны различного размера (шпоровидные клетки) (рис.28).
Рис. 28. Акантоцитоз
94

Этиология синдрома может быть разнообразной: от наследственной абеталипопротеинемии до приобретенных нарушений липопротеинового метаболизма – при печеночной недостаточности, стеаторее, авитаминозе E и голодании.
В группу мембранопатий должна быть переквалифицирова-
на пароксизмальная ночная гемоглобинурия, или болезнь Маркиафава-Микели, обусловленная дефектом мембраны эритроцитов, вследствие чего эритроциты приобретают повышенную чувствительность к действию комплемента. Проявляется ночной гемоглобинурией. Причиной является дефицит гликанинозитолфосфатида – липополисахарида, обеспечивающего абсорбцию ингибиторов комплемента. Предполагается, что вследствие активации системы комплемента альтернативным путем под влиянием ацидоза и гиперкапнии во сне развитивается гемолиз. В патологический процесс вовлекаются гранулоциты и тромбоциты.
Мембранопатии могут иметь приобретенный характер (при авитаминозе E – липидной части).
Гемоглобинопатии (гемоглобинозы) возникают либо вследствие дефицита синтеза глобина, либо его структурной аномальности. Гемоглобин представляет собой гетеродимерный тетрамер, состоящий из двух цепей глобина типа α и двух цепей другого типа (β, γ или δ), соединенных с четырьмя молекулами гема.
К нормальным типам гемоглобина относятся: HbA (α2β2 – основной гемоглобин взрослого, 97%), HbA2 (α2δ2 – минорный гемоглобин взрослого, 2%) и HbF (α2γ2 – фетальный гемоглобин, 1%, рис.29).
Рис. 29. Особенности структуры нормальных типов гемоглобина
95

Известно более 50 видов заболеваний с образованием патологических типов гемоглобинов. Выделяют две основные группы наследственных нарушений образования гемоглобина: одиночные аминокислотные замены в структуре глобина (серповидноклеточная анемия) и уменьшение выработки глобиновых цепей одного или более типов (талассемия или средиземноморская анемия), рис. 30.
Рис. 30. Особенности гетерополимерной структуры гемоглобина, характерные для гемоглобинопатий
Для наследственных гемоглобинопатий характерна фетализация гемоглобина.
Серповидно-клеточная анемия (СКА) – заболевание, свя-
занное с синтезом патологического гемоглобина S (рис. 31). Гемоглобин S образуется в результате точечной мутации в
гене β-цепи, приводящей к замене в 6-м положении глутаминовой кислоты на валин. Наследуется заболевание по типу неполного доминирования. Данная гемоглобинопатия наиболее распространена у лиц, проживающих в «малярийном поясе» (Средиземноморье, Средняя Азия, Америка, Африка, центральная Индия), рис. 32. Гетерозиготы относительно устойчивы к малярийному плазмодию.
96
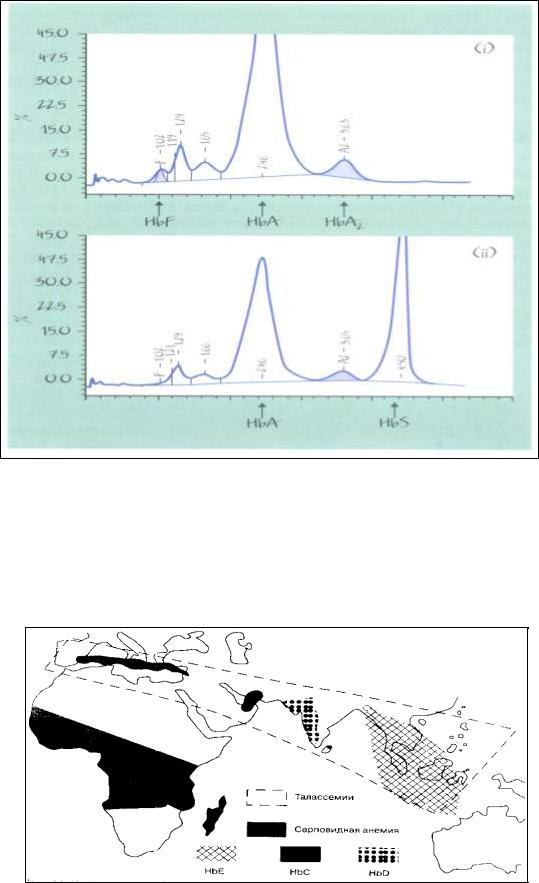
i – Основную часть гемоглобинов крови больного с β-талассемией составляет HbA, но значительно увеличено содержание HbA2 и HbF.
iiHbA и HbS у больного серповидноклеточной анемией.
Рис. 31. Высокоэффективная жидкостная хроматография
Рис. 32. Особенности медицинской географии серповидно-клеточной анемии и других гемоглобинопатий
97

Свойства дезоксигенированных форм гемоглобинов А и S различны в концентрированных растворах: дезоксигенированный гемоглобин S в 50 раз менее растворим, чем HbA. Это лежит в основе временного образования внутри эритроцитов геля и серповидного изменения формы красных клеток при гипоксии (pО2<60 мм рт.ст.), в основном в венозном русле.
Молекулы аномального гемоглобина агрегируют, превращаясь в кристаллы продолговатой формы, изменяя тем самым мембраны и форму эритроцитов (клетки похожи на серп – дрепаноциты) и приводя к гемолизу эритроцитов, рис.33. Средняя продолжительность жизни эритроцитов у больных гомозиготных по гемоглобину S составляет около 17 дней. Деформированные серповидные эритроциты, взаимодействуя с тромбоцитами, эндотелием, коагуляционными белками, вызывают нарушение микроциркуляции вплоть до обтурации сосудов.
Кровь гомозигот содержит до 80% дефектного гемоглобина и в наиболее тяжелых случаях они погибают. У гетерозигот содержание гемоглобина S может варьировать от 30 до 40%, но может достигать 70%.
Рис. 33. Картина крови при серповидноклеточной анемии
Для тяжелой СКА кроме гематологических характерно наличие негематологических острых болевых (окклюзионных) кризов. Их патогенез связан с агрегацией дрепаноцитов в кровенос-
98
ном русле, формированием микроэмболов, ишемией или венозной гиперемией от закупорки соответствующих сосудов и, в конечном итоге – с ишемическими либо венозными инфарктами в различных органах (селезенка, печень, мышцы, ЦНС, кости). Вазоокклюзионные (болевые) приступы могут возникать в области спины, груди, живота, длинных трубчатых костей. Тканевая гипоксия и микроинфаркты могут вызвать острую патологию любой системы организма.
Тромбоэмболы вызывают инсульты, «грудной синдром» – как следствие окклюзии различных ветвей легочной артерии, ишемию и отслойку сетчатки, описаны случаи инфарктов миокарда. Если в циркуляторные нарушения вовлекаются кости и мышцы – криз напоминает «ревматоидный», при поражении внутренних органов – «абдоминальный». Вследствие хронической гипоксии и нарушения текучести крови развивается гиперфункция миокарда и возникает сердечная недостаточность. Тромбофилический синдром является основной причиной гибели этих больных.
По регенераторной способности костного мозга серповидноклеточная анемия является гиперрегенераторной, количество ретикулоцитов составляет около 10% или ≈ 100‰. Подтвердить диагноз может проведение функциональной пробы с наложением венозного жгута на палец.
Талассемия – это широко распростаненная наследственная анемия, для которой характерно снижение или полное отсутствие синтеза α- или β-цепей глобина (соответственно α- или β- талассемия). Компенсаторно усиливается синтез той цепи глобина, генетическая информация которой не изменена. Вследствие несбалансированного накопления α- или β-цепей глобина возникают преципитаты гемоглобина и содержащие их эритроциты удаляются клетками мононуклеарно-фагоцитарной системы. Возникает гипохромная микроцитарная анемия.
Причина α-талассемии обусловлена дефектом интронов α- цепей гемоглобина в хромосоме 16, что ведет к полному либо частичному нарушению их биосинтеза. Эти цепи заменяются в
99
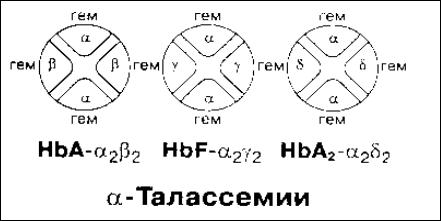
Рис. 34. Особенности гетерополимерной структуры Hb, характерные для талассемий.
пренатальный период на γ-цепи (Hb Барт). В постнатальном периоде происходит замена на β-цепи и возникает тетрамер-β: гемоглобин Н.
Выделяют следующие формы α-талассемии: thalassemia major (при наличии 80-90% Hb Барт), thalassemia intermedia c 60% HbH и 40% Hb Барт, thalassemia minor – не более 5% Hb Барт, thalassemia minina – без клинических проявлений с уровнем Hb Барт не более 2%. Thalassemia major приводит к гибели в фетальный или ранний постнатальный период, в то время как thalassemia major не имеет клинических проявлений.
Клиника определяется гипоксией из-за плохой диссоциации HbО2, а также укорочением срока жизни эритроцитов.
При β-талассемиях происходит нарушение синтеза β-цепей гемоглобина вследствие мутирования интронов в 11-й хромосоме и компенсаторное усиление синтеза α-цепей.
При гемоглобинопатиях и мембранопатиях преобладает внутриклеточный гемолиз. При данных видах гемолитических анемий уменьшается деформируемость эритроцитов, т.е. они становятся ригидными, малоэластичными. Проходя с трудом по микроциркуляторному руслу, они повреждаются, захватываются макрофагами селезенки и печени и подвергаются преждевременному гемолизу.
При некоторых гемоглобинопатиях вследствие окисления глобина в составе молекул нестабильных гемглобинов образуются особые внутриэритроцитарные включения (преципитаты гемоглобина) – тельца Гейнца. Образованию окисленных преципитатов способствуют нарушения энергетического обмена, приводя-
100