

Люпус-нефрит
.pdfSection 5 Glomerular Disease
Chapter
Lupus Nephritis
27 J Stewart Cameron
INTRODUCTION AND DEFINITION
The importance of lupus nephritis1–4 to practicing nephrologists is that it is a serious but usually treatable disease. The term ‘lupus’ (Latin: wolf) has been used for many centuries to denote any skin condition where one component is ulceration. Although Brooke first noted albuminuria in lupus in 1895, recognition of visceral involvement including renal disease was slow for the next decade or two. The modern era began in 1957 with the amazingly complete description by Kark, Muehrcke and colleagues of renal biopsy appearances of lupus nephritis5.
Lupus is defined by its clinical picture, together with antibodies directed against one or more nuclear components, particularly double-stranded DNA6. It is best regarded as a syndrome in which a number of different patterns of immune disturbance may lead to a similar final common pathway and thus disorders with similar clinical pictures. The criteria of the American College of Rheumatology (ACR), revised in 1982 and modified slightly in 1997, have been applied widely to the diagnosis of lupus and have proved durable – although they were introduced originally only to distinguish the disease from other closely related clinical conditions (Table 27.1). The presence of four or more of the major criteria is usually taken as establishing the diagnosis with about 96% sensitivity and specificity.
Defining ‘typical’ or ‘core’ patients in this fashion excludes a considerable number of patients with ‘lupus-like’ disorders who should also be recognized and treated. Many patients with a clinical lupus syndrome but a negative antinuclear antibody (ANA) have low titers of anti-Ro antibody and rarely have significant renal disease, but often show a high incidence of antiphospholipid antibodies (APA) with associated thromboses and abortions, as well as inherited complement deficiencies.
ETIOLOGY AND PATHOGENESIS
Origins of autoimmunity
How autoimmunity and the lupus syndrome arise remains obstinately obscure7,8. In lupus there is a generalized autoimmunity, with autoantibodies directed against a variety of self components, but the role of the autoantibodies in generating organ damage is unclear. This contrasts with organ-specific autoimmunity (such as antiglomerular basement membrane (anti-GBM) nephritis), in which clearly pathogenetic autoantibodies are directed against a single self epitope.
Genetic factors
Genetic factors are important in lupus, with racial preponderance and familial clustering, and monozygotic twins showing 25% concordance. In addition, healthy relatives of patients with lupus may show antinuclear and other autoantibodies. Only weak major histocompatibility complex (MHC) associations have been noted, the strongest being with C4A or C4B null genes and low tumor necrosis factor (TNF) production. A few patients may show genetic deficiencies of complement components, and some studies have shown associations with Fcγ RIIIa receptor polymorphisms. Acquired complement deficiencies occur also, for example in complement receptor CR1 and a pathogenic role for poor immune clearance of complexes has been postulated. However in human lupus there is no evidence for genetic defects in the apolipoprotein-1 (APO- 1)/fas receptor whose engagement leads to apoptosis, such as has been described in some forms of murine lupus.
Other etiologic agents
There is no convincing evidence that infective agents provoke human lupus. Hydralazine, procainamide, and occasionally other drugs, may precipitate a lupus syndrome which, however, rarely affects the kidney. Interaction of genetic and
The American College of Rheumatology criteria for the diagnosis of lupus
The presence of four or more of the following criteria gives 96% sensitivity and specificity for the diagnosis of lupus:
1.Malar rash
2.Discoid rash
3.Photosensitivity
4.Oral ulcers
5.Nonerosive arthritis
6.Pleuropericarditis
7.Renal disease (proteinuria and/or granular casts)
8.Neurologic disorder (fits or psychosis in the absenceof precipitating circumstances)
9.Hematologic disorder (hemolytic anemia,leukopenia/lymphopenia, thrombocytopenia)
10.Positive LE cell preparation, raised antiDNA antibody, anti-Sm present, false-positive antitreponemal test
11.Positive, fluorescent, antinuclear antibody test
Table 27.1 The American College of Rheumatology criteria for the diagnosis of lupus, revised 1982 and modified in 1997.
357 |
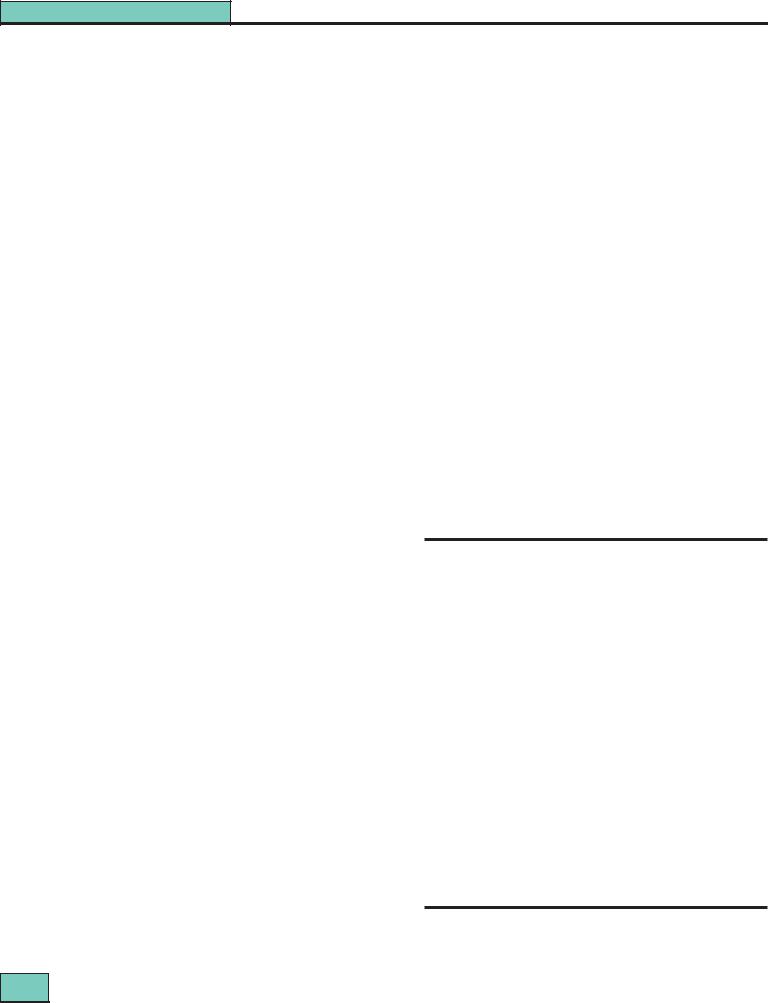
Section 5
Glomerular Disease
environmental factors is shown in hydralazine-induced lupus: three genetic factors (female gender, slow acetylator status, and HLA-DR4) plus exposure to hydralazine account for 98% of the risk.
Animal models of lupus nephritis
Spontaneous lupus has been reported in a number of strains of mice, such as the NZB B/W F1 hybrid and MRL-lpr. Both have primary defects that lead to B-cell proliferation, including defects in the fas/APO-1-ligand system, which has a role in apoptosis. It is assumed in these animals that defective apoptosis leads to defective clonal deletion. In addition, lupus can be provoked in mice by injecting autoantibodies against DNA or phospholipid, or by inducing graft versus host disease, and in rabbits by injection of peptides derived from the Sm-antigen.
Pathogenesis
Typically there are multiple autoantibodies in lupus directed against nucleic acids and proteins concerned with intracellular transcriptional and translational machinery6. The main targets are nucleosomes (DNA-histone) or even quaternary antigens on the chromatin itself, small nuclear ribonucleoproteins (snRNPs), and small cytoplasmic RNPs (scRNPs).
Polyclonal hyperactivity of the B-cell system or defects of T-cell autoregulation are likely primary events in lupus. One hypothesis is that some autoreactive T cells survive thymic deletion and persist into adult life in a state of suppression, with the emergence of clones of autoreactive cells and antibodies and tissue damage if this suppression fails. A second hypothesis is that presentation of self antigen (such as his- tone-derived peptides) to a mature immune system is capable of inducing germline mutations resulting in the production of new autoantibodies, that are not adequately suppressed. A variant of this hypothesis is that viral or bacterial peptides contain sequences that are similar or identical to those of native antigens with which they cross-react: so-called ‘antigenic mimicry’. A third hypothesis is that there is nonspecific, polyclonal B-cell stimulation via superantigens, the resultant B-cell repertoire including pathogenetic autoantibodies that again fail to be suppressed.
Mediation of tissue injury
Patients with lupus nephritis most usually show antibodies directed against dsDNA, Sm, and C1q. But it has proved difficult to show that the DNA–antiDNA antibody system, so characteristic of lupus, has a direct role in pathogenesis.
Aggregates of immunoglobulin and complement components are present at sites of injury in the glomeruli, and in the tubules in about two-thirds of renal biopsies. Whether these are derived from circulating complexes or from in situ combination of antigen and antibody is still unclear. Deficiencies in the handling of immune complexes and other foreign material have been described. These are perhaps inherited in association with the MHC haplotype HLA-A1-B8-DR3 or Fcγ RIIIa receptor polymorphisms.
Anti-dsDNA antibody has been eluted from nephritic kidneys together with dsDNA and histone, but the mere presence of antibody does not prove it to be damaging. Infusions and
transplacental transmission of antiDNA antibodies do not lead
358
to nephritis in humans. Only in models in experimental animals is there direct evidence that murine or human antiDNA antibodies can penetrate cells and cause proteinuria.
In some instances dsDNA–anti-dsDNA antibody complexes fix to DNA receptors on cells, including endothelial cells. In others histones appear to mediate binding to both matrix and cells. Histones are present at sites of immune aggregates in murine and human lupus nephritis, but not in primary glomerulonephritis.
There is no reason to believe that the effector mechanisms of renal damage in lupus are different from those of primary glomerulonephritis. However the interstitial cellular infiltrates in lupus have an excess of CD8+ cytotoxic T lymphocytes over CD4+ compared to the usual mix of CD4+ helper T lymphocytes and monocytes seen in primary glomerulonephritis.
Immunologic findings in patients with and without nephritis
Why do only some patients with lupus develop clinically evident nephritis? We do not have an answer to this question, but those with nephritis usually have antibodies directed against dsDNA as well as ssDNA, and have at most low titers of anti-Ro and anti-La antibody. They also have high-avidity anti-DNA antibodies that activate complement strongly. Higher-avidity antiDNA antibodies also occur in proliferative more than in membranous lupus nephritis and cationic antibodies appear to be more pathogenetic. Antibodies directed against C1q are also more frequent in those with nephritis.
EPIDEMIOLOGY
Prevalence
Incidence, prevalence and mortality1,4 are ten times higher in African American females (prevalence ~1 : 400) than in Caucasians, whereas it appears relatively rare in West Africans – except perhaps in urban areas. In the USA lupus is twice as frequent in Orientals compared to Caucasians, although there is a relatively low prevalence in mainland China, Taiwan, and Japan. On the other hand, lupus seems to be extremely common in the relatively small Chinese populations of both Singapore and Hong Kong. It is also more common in South Asians. There is some evidence that lupus and its associated nephritis are becoming more common.
Gender and age
Gender is the major risk factor for the development of lupus. The female : male ratio rises from 2 : 1 in prepubertal children up to 4.5 : 1 in adolescents to 8–12 : 1 in adults, falling back to 2 : 1 in patients over 60 years of age. These data are in accord with murine models of lupus where estrogens are precipitating factors in the emergence of lupus, whereas androgens protect. Lupus is rare before puberty.
CLINICAL MANIFESTATIONS
Renal manifestations of lupus9
Only 30–50% of unselected patients with lupus have abnormalities of urine or renal function early in their course2,9, but up to 60% of adults and 80% of children may develop overt

Chapter 27
renal abnormalities later. Although in those with onset at more than 50 years of age, nephritis is distinctly less common (< 5% at onset).
The dominant feature in almost every patient with renal lupus is proteinuria (Table 27.2)9. Surprisingly, hypertension is not, overall, more common in those with nephritis but those with more severe nephritis are more commonly hypertensive. Renal tubular function is disturbed, which is not surprising in view of the findings of both immune aggregates in tubular basement membranes and interstitial nephritis. In a high proportion of patients, urinary excretion of light chains and β 2- microglobulin are both increased. Recently distal renal tubular acidosis has been emphasized as a manifestation of lupus. Bladder involvement may be prominent, often a severe immune interstitial cystitis10.
Extrarenal manifestations of lupus
Overall those patients with lupus nephritis tend to have more alopecia and oral ulceration than those without, but have less arthritis, facial rash, and Raynaud’s phenomenon2. The initial complaints are often nonspecific, with three-quarters of patients showing fever and malaise without weight loss.
A rash occurs in half to three-quarters of cases, usually the well-known ‘butterfly’ rash on the face; livedo reticularis may be seen on exposed areas in up to 15% of cases and may be associated with APA. The rash may be vasculitic with alterations in the nailbed capillaries and sometimes ulcerating lesions, especially around the ankles. Discoid lupus is unusual in patients with lupus nephritis, but photosensitivity is common. Some degree of hair loss is common, which amounts to patchy alopecia in a few cases. Oral ulceration is a presenting feature in 10% of patients.
Raynaud’s phenomenon is common in young adults with lupus, affecting 20–30% and often preceding the clinical onset of other manifestations of disease. In some patients it is very severe with loss of digital tissue, as in scleroderma. Patients with renal disease rarely have severe Raynaud’s, however, and overall it is a favorable feature in terms of survival.
The arthralgia of lupus is common, occurring in threequarters of patients and is almost never deforming. Usually several joints are affected at once, often in the hands, but almost any joint may be a target; some myalgia is common in untreated patients at onset, often accompanied by weakness, but myositis is rare.
Neuropsychiatric involvement is one of the most serious extra-renal manifestations of lupus11 and may be more common and run a more severe course in Orientals, and perhaps also African Caribbeans. It is apparent clinically in about one-third of patients, and is a presenting feature in about 10%. Mood and behavior disorders of a minor degree are common. They are difficult to interpret in the setting of an acute and disturbing illness but, especially if associated with persistent headache (sometimes migrainous), may be the prodrome of serious, overt, neuropsychiatric disorder.
Chorea may be seen, especially in children with neurologic lupus, sometimes in association with APA. In addition, cranial nerve palsies, for example ophthalmoplegias, brainstem lesions and hemiparesis may occur in addition to coma and frank psychosis. Cerebral bloodflow studies, positron emission
Lupus Nephritis
Clinical features of patients with evident lupus nephritis
|
% |
|
|
Proteinuria |
100 |
Nephrotic syndrome |
45–65 |
Granular casts |
30 |
red-cell casts |
10 |
Microscopic hematuria |
80 |
Macroscopic hematuria |
1–2 |
Reduced renal function |
40–80 |
Rapidly declining renal function |
30 |
Acute renal failure |
1–2 |
Hypertension |
15–50 |
Hyperkalemia |
15 |
Tubular abnormalities* |
60–80 |
* usually without symptoms
Table 27.2 Clinical features of patients with evident lupus nephritis
tomography scanning, and magnetic resonance imaging may show abnormalities additional to those revealed by computerized tomography, even in patients without obvious neuropsychiatric symptoms, but their significance is not yet clear.
Pleuritis and pericarditis affect about 40% of patients; these disorders are usually painful but sometimes symptomless effusions develop. Pericarditis with heart failure does occur but is rare. Endocarditis of the Libman–Sacks type has been associated by some with the presence of APA. It is often symptomless and difficult to diagnose except by echocardiography.
Pulmonary hypertension in lupus may be the result of multiple pulmonary emboli in association with APA, sometimes with vena caval thrombosis. However, it is associated clinically with Raynaud’s phenomenon in about three-quarters of cases and may represent a similar vasospastic phenomenon in the lung. Treatment is ineffective and the outlook poor: heart–lung transplantation is possible in some cases. During the acute phase, as well as respiratory infections, acute, potentially fatal, pulmonary hemorrhage may be seen, albeit rarely. Acute, reversible hypoxemia is common, the pathogenesis of which is unclear. Chronic fibrosing alveolitis, a wellrecognized feature of lupus, may be progressive and treatment is unsatisfactory.
Splenomegaly and lymphadenopathy are present in about one-quarter of patients with lupus, often with fever and weight loss although this group rarely develops severe nephritis.
Clinical hematologic abnormalities are common. Many patients have a normochromic, normocytic anemia at presentation. Occasional patients present with purpura, not from associated vasculitis but from thrombocytopenia. Thromboses present in about 12% of patients and their occurrence should prompt a search for APA and other procoagulant abnormalities. If a patient with lupus develops a nephrotic syndrome, there will be the additional thrombogenic potential of the alterations in platelet function and plasma coagulation factors that are seen in all nephrotics.
Thrombosis occurs in 5–15% of patients with lupus nephri-
tis and may affect almost any vessel in the body. Venous
359

Section 5
Glomerular Disease
thromboses occur most frequently, if an APA is present. Arterial thromboses are also seen, cerebral thrombosis being particularly common. Inferior vena caval thrombosis may be seen, and pulmonary embolism is common, perhaps causing pulmonary hypertension. Unilateral or bilateral renal venous thrombosis is common also, especially in lupus membranous nephropathy (WHO class V). A number of patients with a mixed picture of lupus and thrombotic microangiopathy have been described who may develop acute renal failure and who in general have a poor prognosis.
Laboratory investigations in lupus nephritis
Antinuclear antibodies
Antinuclear antibodies6, particularly those against doublestranded DNA (present in up to 90% of untreated lupus) and the Smith (Sm) antigen, are strongly associated with the presence of nephritis. The Smith (anti-Sm) antibody is almost pathognomonic for the diagnosis of lupus. It is highly specific, but present only in some 30% of patients with nephritis, more in African-Caribbean patients than in Caucasians. Treatment may rapidly eliminate anti-dsDNA antibodies from the circulation while the fluorescent antinuclear antibody (FANA) test remains positive. The various patterns of FANA (diffuse, speckled etc.) are not reliable in distinguishing lupus from other ANA-positive diseases.
Hematology
Anemia of moderate degree is common, but a positive test for anti-red-cell antibodies (Coombs’ test) can be obtained only in a minority of patients with lupus, and severe hemolytic anemias are not often seen. Leukopenia (caused by anti- white-cell antibodies) is common, and 50% of patients have a white-cell count below 5 × 109/L, while thrombocytopenia is found in about one-quarter of patients. The origins of the thrombocytopenia are complicated, resulting from accelerated platelet destruction after binding of antiplatelet antibody or APA and/or lysis after phagocytosis of circulating immune complexes.
Antiphospholipid antibodies and the ‘lupus anticoagulant’
The double misnomer ‘lupus anticoagulant’ activity12 is based upon the presence of APA, directed mainly against the β 2- globulin phospholipid-carrier-protein. These antibodies prolong phospholipid-dependent coagulation studies in vitro (activated partial thromboplastin time (APTT) and kaolin clotting time (KCT)) but in vivo are associated with thrombosis. The prolonged APTT and KCT are not corrected by mixing with normal plasma. The in vitro mechanisms are clear but the reason for thrombosis in vivo remains uncertain. APA can be detected in about one third to one half of patients with lupus nephritis, and have been associated with renal arterial, venous, and glomerular capillary thrombosis, as well as Libman–Sacks endocarditis and cerebral thrombosis. Prothrombotic risk factors other than APA include depressed release of plasminogen activator and possibly also of antagonists of plasmin, decreased plasma concentration of free protein-S, and raised von Willebrand factor concentrations.
As well as APA, true lupus anticoagulants may be present in
the form of antibodies directed against factors leading to fibrin
360
formation, such as factor VIII and IX, but also less commonly factors XI and XII. These lead to clinical bleeding as well as prolongation of clotting times, which are corrected on mixing with normal plasma.
It is important to note that despite the in vitro prolongation of clotting times, it is safe to do needle biopsies in the presence of APA. In contrast, prolongation of KCT, which reverses on mixing with normal plasma, is the action of a true anticoagulant and will require cover with fresh-frozen plasma.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The diagnosis of lupus is usually easy but may, on occasion, be very difficult, especially in more unusual circumstances such as a middle-aged, nephrotic male or apparently idiopathic membranous nephropathy in a young woman. Thus it should be routine to screen all proteinuric patients for anti-nuclear antibodies. About 50% of patients with lupus are initially suspected of having a disease other than lupus, most commonly rheumatic fever, rheumatoid arthritis, and hemolytic anemia. The presence of four or more of the ARA criteria has a 96% sensitivity and specificity when applied to patients seen in rheumatology clinics where the criteria were originally defined. Differentiation from rheumatic fever is relatively easy, but in a child with chorea it may not be straightforward.
Nephritis has been reported in a minority of patients with mixed connective tissue disease; the differential diagnosis can be difficult clinically, but analysis of the antinuclear antibody for the anti-Ro and anti-La antibodies and the absence of anti-dsDNA antibodies should make the diagnosis clear.
Rheumatoid arthritis usually does not show systemic features but on occasion, proteinuria will be induced by one of the drugs used in its treatment and cause additional problems in diagnosis. Some of these patients go on to develop full clinical and immunologic lupus. The presence of erosions and a deforming arthritis makes lupus very unlikely, but does not exclude it.
Henoch–Schönlein purpura is much commoner in childhood than lupus, and on occasion differentiation may be difficult since the rash of lupus may be purpuric and affect the lower limbs only. A few patients with lupus may have IgA predominant in their renal biopsies with raised serum IgA concentrations. Lupus may on occasion be complicated by a vasculitis, which creates difficulties in differentiating it from other forms of vasculitis, especially when p-ANCA (antineutrophil cytoplasmic antibodies may seem to be present.
Immunologic tests and the diagnosis of lupus
Few clinicians are happy to make a diagnosis of lupus nephritis without some antinuclear antibodies in the serum, preferably those shown to react with dsDNA. ‘Lupus-like’ patients with negative antinuclear antibody tests generally present with little or no renal disease, although there are exceptions, and more than 80% of this ‘fringe’ have APA.
The proportion of positive ANA depends not only upon the population studied but upon the technique used6. The classic Farr assay detects only high-avidity anti-dsDNA antibodies; enzyme-linked immunosorbent assay (ELISA) also picks up

Chapter 27
low-avidity antibodies as does the Crithidia lucilae kinetoplast slide test. Correlations with the presence and severity of nephritis are best with high-avidity antibodies using the Farr assay, but for screening diagnosis, the ELISA assay has advantages since it will detect positives in some FANA-positive patients in whom the Farr assay is negative, but who do have lupus. Anti-Sm antibodies are almost entirely specific for lupus, but are found only in about 30% of patients and thus have a very low sensitivity.
Hypocomplementemia is found at presentation in more than three quarters of untreated, younger patients with lupus and is more common with evident nephritis. C4 and C1q tend to be more depressed than C3 suggesting complement activation via the classical pathway; this pattern is rare in membranoproliferative GN (MPGN) type I and poststreptococcal GN, but is common in MPGN type II and essential cryoglobulinemia (see Chapter 23). However, concentrations of properdin and factor B are also depressed, with activation of the alternative pathway. The C5b-9 complex is also found in the circulation in increased amounts. In the absence of active disease null C4 genes will confer concentrations of about half normal C4 in heterozygotes, and absent C4 in homozygotes.
The interpretation of antineutrophil cytoplasmic antibodies (ANCA) is difficult in the presence of antinuclear antibodies. However, the finding of multiple immunoglobulin deposition together with complement in the affected glomeruli and a proliferative/membranous pattern rather than a pauci-immune necrotizing glomerulonephritis, should cause no diagnostic difficulty.
Immune complexes can be detected in the serum of the majority of patients with lupus, especially those with nephritis, and the titer generally rises and falls with indices of clinical activity. However, their clinical utility is minimal since immune complexes can be detected in so many other conditions, and immune-complex detection is no longer in routine clinical use. Complement-based tests have been shown to measure not immune complexes, but anti-C1q autoantibodies.
Lupus Nephritis
PATHOLOGY3,13
Renal biopsy is worthwhile in all patients with lupus and abnormal urine. It provides prognostic information and influences initial treatment. The overriding characteristic of lupus nephritis is its variability between patients, within biopsies, and even within glomeruli.
Glomerular appearances
The World Health Organization (WHO) classification of lupus nephritis (see Figs 27.1–27.5), which is based on light microscopy, is widely accepted but allows only a broad judgment of severity. Class III (focal proliferative nephritis) is a particular source of difficulties since it covers such a wide range of appearances. Nevertheless, there is a remarkable similarity in the proportion of patients allocated to each class in different series; more than half show WHO class III (focal proliferative) or IV (diffuse proliferative) nephritis – severe forms that most clinicians would treat vigorously. The proportion of class V (membranous) biopsies is about the same in all series (10–15%).
On immunohistology, IgG is almost always the dominant immunoglobulin, IgG1 and IgG3 being especially prevalent, but a few patients have predominant IgA or IgM. Early complement components such as C4 and especially C1q are usually present along with C3. The finding of positivity for all three isotypes of immunoglobulin together with C3, C4, and C1q is called a ‘full house’ and is present in about one-quarter of patients with lupus and almost never in non-lupus disease. Other immune reactants such as complement components H, B, C5b-9, and properdin are present also in many patients. Fibrin, sometimes accompanied by cross-linked fibrin, is often present in class IV biopsies but rare in other classes.
Tubulointerstitial nephritis
In about 50% of patients with nephritis, less in those with class II and up to three-quarters of those with class IV,
Figure 27.1 WHO class I lupus nephritis (minimal changes): present in <5% of biopsies (dependent upon biopsy policy in patients with mild or no renal disease). Light microscopy is normal, but immunoperoxidase shows extensive C1q (associated with IgG and C3) throughout the mesangial areas.
a
Figure 27.2 WHO class II lupus nephritis (mesangial disease): present in 10–25% of renal biopsies. (a) Mesangial expansion but little increase in tuft cellularity and the peripheral capillary walls are normal. (Silver methenamine stain.) (b) Extensive mesangial IgG deposits shown by immunoperoxidase; the aggregates are just beginning to invade peripheral capillary walls. This represents the most severe changes in this class.
361 |
Section 5
Glomerular Disease
a |
b |
c |
Figure 27.3 WHO class III lupus nephritis (focal proliferative): present in 20–35% of renal biopsies. (a) An area of focal necrosis containing cellular debris (arrow) is surrounded by a degree of epithelial cell proliferation (silver methenamine/hematoxylin eosin.) This is a mild example of class III nephritis.
(b) A more severe example of class III: numerous areas of the tuft are affected by segmental capillary wall thickening and mesangial expansion, with cellular proliferation and infiltration. (PAS.) (c) A major focal and segmental lesion affecting almost half the glomerular tuft: another severe variant of class III (hematoxylin/lissamine green).
|
|
D |
D D |
|
|
D |
|
|
|
D |
|
|
|
D |
|
a |
b |
D |
M |
c |
|
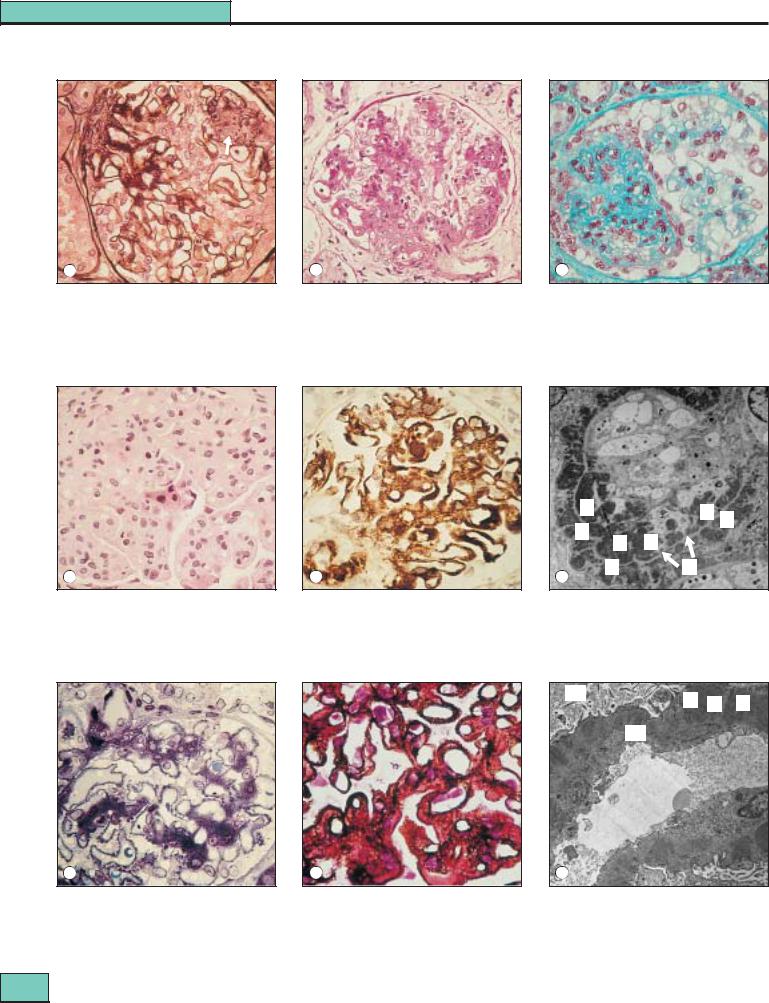
Figure 27.4 WHO class IV lupus nephritis (diffuse proliferative): present in 35–60% of biopsies. (a) The tuft is increased in size by both a diffuse increase in matrix and an excess of cells; capillary walls are irregularly thickened (hematoxylin/eosin). (b) Dense, irregular aggregates of IgG along the peripheral capillary walls by immunoperoxidase. (c) Electron microscopy shows the immune aggregates as electron-dense masses (D) at both subendothelial and subepithelial sites, with the lighter capillary basement membrane (M) (arrows) between them.
US
D S D
BMBM
a |
b |
c |
Figure 27.5 WHO class V (membranous): present in 10–15% of biopsies. (a) The predominant aggregates are along the outside of the capillary wall. Note that there are also abundant dark-blue, mesangial aggregates, as well as those along the capillary wall (class Vb). Focal and diffuse proliferative lesions are absent. (b) Silver methenamine staining shows the silver positive ‘spikes’ along the peripheral capillary wall protruding between the immune aggregates. (c) Electron microscopy of a capillary loop and adjacent urinary space (US) shows electron-dense immune aggregates (D) on the outer surface of the basement membrane (BM), separated by ‘spikes’ (S) of exuberant basement membrane-like material, mainly laminin. The immune aggregates in
class V membranous lupus nephritis are typically more irregular in size and position than those commonly found in idiopathic membranous nephropathy.
362

Chapter 27
Lupus Nephritis
a
b
Figure 27.6 Interstitial lupus nephritis. (a) Interstitial infiltrate invading and destroying tubules (‘tubulitis’). The tubular basement membrane (TBM), stained black with silver (arrow), is digested (see the lower half of figure(a)) and lymphocytes together with macrophages invade the tubules. (Silver methenamine/hematoxylin.) (b) Immunofluorescence showing aggregates of C3 in the TBM (right) as well as within the glomerulus (left). Such TBM aggregates are common in lupus nephritis, being found in 60–65% of biopsies overall and with increasing frequency from class II (20%) through to class IV (75%).
immune aggregates are present in the tubular basement membrane (TBM). In an occasional patient, linear tubular immunofluorescence is seen, suggestive of anti-TBM antibodies. The interstitial infiltrate is mainly T lymphocytes (both CD4+ and CD8+) and monocytes, with only a few B cells, plasma cells, and natural killer cells. Among the T lymphocytes, are present. Infiltration and invasion of tubules (tubulitis) is frequently present (see Fig. 27.6)14 in active disease. In more chronic disease the interstitium is expanded with variable amounts of collagen. In a few patients an acute tubulointerstitial nephritis is seen in the absence of glomerular disease and may present as acute renal failure.
Intrarenal vessels
Vascular immune aggregates, hyaline, and non-inflammatory necrotizing lesions, and true vasculitis with lymphocytic and monocyte infiltration of the vessel wall, may all be seen and,
Figure 27.7 Vascular damage in lupus nephritis. ‘Thrombus’ (arrow) occludes a glomerular capillary loop in this class IV biopsy. Such a ‘thrombus’ contains platelets and cross-linked fibrin as well as immunoglobulins and thus has some characteristics of true thrombus. Note also the subepithelial aggregates, spike formation, and ‘double contouring’ of the capillary walls, all typical of class IV and active class III biopsies. (Silver methenamine/hematoxylin.)
more rarely, intrarenal arteriolar thrombi (see Fig. 27.7)15. All these vascular changes are signs of a poor prognosis. Occasionally, patients show overt thrombotic microangiopathy. Correlations have been shown between the presence of APA and intraglomerular thrombi.
Other glomerular appearances
Amyloidosis is rare in lupus. This is consistent with the fact that acute phase proteins such as amyloid-A and C-reactive protein do not rise during acute flares of activity in the disease. Dense deposit disease has occasionally been reported in the context of lupus.
Transformation of histologic appearances
Serial biopsies show that transformation of WHO classes is quite frequent. A particularly common transformation is one from diffuse proliferative glomerulonephritis (class IV) to a predominant membranous (class V) pattern under successful treatment. Under these circumstances, proteinuria may become massive as renal function improves.
Inapparent renal disease in lupus
Patients without clinical manifestations of nephritis may have significant glomerular disease on renal biopsy. There have been few follow-up studies on such patients, but one study showed that the majority remain without clinical nephritis for some years. However, it is equally obvious that all patients with clinically evident nephritis must go through a period of absent or occult disease before the disease becomes evident; how many of these patients run a subclinical course for a prolonged period is not known.
Clinicopathologic correlations
While nephritis that is histologically more severe has a ten-
dency to more severe clinical manifestations, renal histology
363
Section 5
Glomerular Disease
Clinicopathologic correlations in lupus nephritis
|
WHO class II |
|
WHO class IV |
|
WHO class III |
|
WHO class V |
|
|
100
80
60
%
40
20
0
Nephrotic |
Proteinuria |
Normal |
Diminished |
syndrome |
|
renal |
renal |
|
|
function |
function |
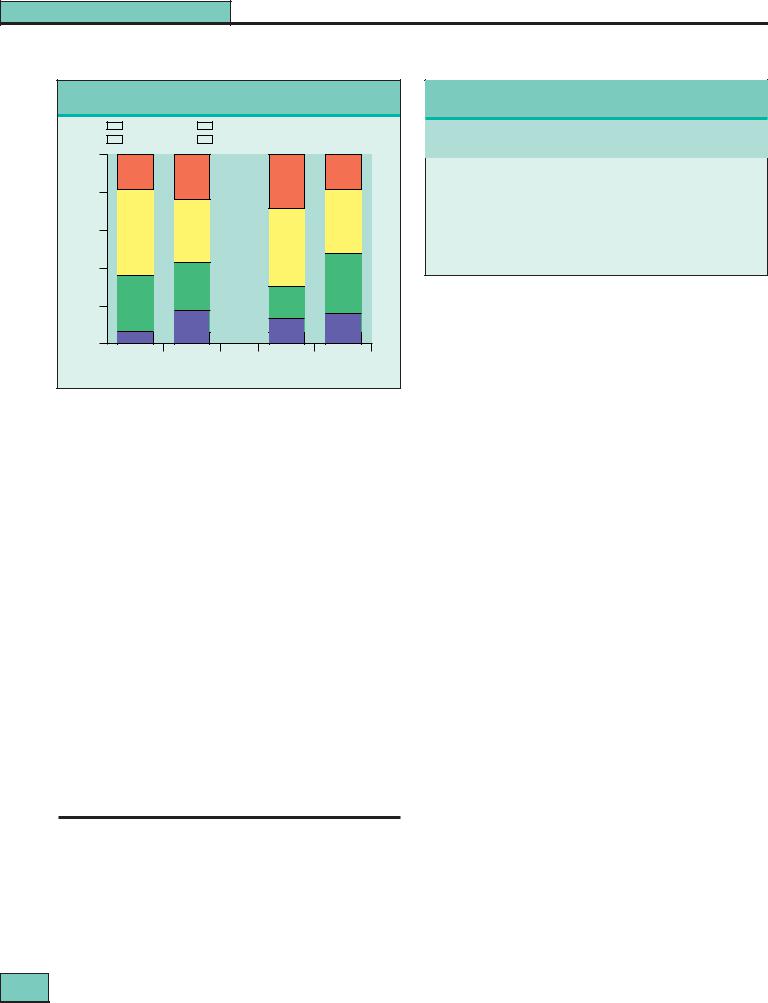
Figure 27.8 Clinicopathologic correlations in lupus nephritis.
Correlations between the presence of a nephrotic syndrome, proteinuria, and normal or diminished renal function with the WHO class on renal biopsy. The clinical picture has almost no value in predicting the biopsy appearances.
cannot be predicted with any certainty from the clinical picture (see Fig. 27.8). In untreated patients, WHO biopsy class is a powerful determinant of outcome. However, this is no longer true if patients with more severe nephritis are given more active treatment (see below). Interstitial changes (both in cells and interstitial volume) correlate well with glomerular filtration rate (GFR) at the time of biopsy, as well as with outcome. Titers of anti-dsDNA antibody were similar in our own patients in all histologic groups, but other researchers have noted significant, though not clinically useful, differences. By the time a renal biopsy is done the patient will almost certainly have received some immunosuppressive treatment, which will alter the serologic results.
The group at the US National Institutes of Health (NIH) have popularized a histologic scoring system for disease ‘activity’ versus ‘chronicity’ to generate a numeric assessment of the biopsies. Others, however, have been unable to replicate its prognostic utility.
NATURAL HISTORY
Outcome and mortality in treated patients in recent years
The clinical course of lupus can no longer be considered separately from the results of treatment. Forty or 50 years ago, few patients with severe grade IV nephritis survived more than a year or two, and half of those with even less severe forms of nephritis used to die within 5 years (see Table 27.3). There has now been marked improvement in the outcome for patients with class IV biopsies, which today differs little from lupus
nephritis as a whole.
364
Table 27.3 Survival in lupus and lupus nephritus
|
5 Year survival (weighted mean of published series) % |
||||
Period |
|
All lupus |
Lupus nephritis |
Class IV nephritis |
|
|
|
|
|
|
|
1953–1969 |
49 |
44 |
17 |
|
|
1970–1979 |
82 |
67 |
55 |
|
|
1980–1989 |
86 |
82 |
80 |
|
|
1990–1995 |
92 |
82 |
82 |
|
|
|
|
|
|
|
|
Five-year actuarial survival for lupus, lupus nephritis, and WHO class IV nephritis over the past 40 years.
Table 27.3 Survival in lupus and lupus nephritus
There seems little doubt that this improvement has been the result of better management of lupus nephritis and this has led to almost all patients with the disease receiving treatment, including those with the mildest forms. However, the early data of Ropes16 must not be forgotten: in the 1960s she studied 68 unbiopsied patients with lupus and proteinuria (some nephrotic) who were left without treatment, and in 16 (28%) cases the proteinuria became intermittent or disappeared spontaneously.
In most patients today, there is a gratifying response to early treatment, followed by relatively quiescent disease under continuing immunosuppression that can be tapered off eventually without further relapse. Another common pattern is the quiescent patient who suddenly relapses. The frequency of relapse depends not only on the underlying disease but on the intensity and duration of immunosuppression.
Since lupus is a multisystem disease, the outlook does not of course depend only upon what happens to renal function, especially now that this is treatable by dialysis and transplantation. Also there are many accessory factors such as the socioeconomic circumstances of the family and the comprehension of treatment goals by the patient. These goals must be adhered to over many years, even when the patient is apparently well but perhaps suffers side-effects of treatment. Measures to deal with these psychosocial problems may be as important as specific treatments in determining survival.
Although long-term results are now very encouraging compared with 20 years ago (Table 27.3), many questions remain unresolved.
•Will reduced renal function evolve eventually into renal failure?
•What is the significance of persistent proteinuria in the absence of disease activity?
•How long should treatment continue and what should it be?
•On what grounds can treatment be stopped safely?
The causes of death in lupus are much more varied than in primary glomerulonephritis, in which renal failure is the dominant cause. End-stage renal disease (ESRD) now affects 8–15% of patients with lupus nephritis, and fatal infections are the usual cause of death in this group. Infection also remains the commonest reported cause of early death in

Chapter 27
those without ESRD, although retrospective reports may not reflect current practice. Extra-renal disease is the next most common cause of early death, particularly affecting the CNS or lung. But overall almost half of all lupus deaths are the result of excess cardiovascular mortality, often later in the course of the disease and particularly from premature myocardial ischemia.
Outcome in relation to clinical, histologic, and laboratory findings
There has been more controversy than agreement as to which features might be associated with a poorer outcome in lupus nephritis1,3,17,18. Factors influencing outcome in some series but not others are: age at onset of > 55 years, childhood onset, Black race, raised plasma creatinine, and hypertension. Indices of clinical activity, number of ACR criteria present at onset and the number of clinical relapses also predict outcome.
Today there is little difference in outcome between different WHO classes of nephritis in treated patients, although extensive subendothelial deposits and tubulointerstitial changes point to poorer prognosis as do the number of macrophages and T cells in the interstitium. Crescents have also been related to a poorer prognosis, as in other forms of nephritis, but extensive crescentic disease is uncommon in lupus.
Vascular lesions within the biopsy and intraglomerular capillary thrombi have been associated with unfavorable outcomes also, although the latter observation has been contested. Calculation of activity and chronicity indices has allowed the NIH group to identify groups of high and low risk for a poor outcome and also has permitted therapeutic decisions, especially when and when not to use aggressive treatment although these data are not supported by all other analyses.
Correlations between complement concentrations and levels of circulating anti-dsDNA antibody and outcome have been reported but are not useful in practice. The principal useful predictor amongst laboratory tests is anemia, with thrombocytopenia, hypocomplementemia, and a raised DNAbinding at onset all correlated with a poorer outcome.
TREATMENT19–39
Despite a half-century of clinical inquiry, the useful data upon which to base recommendations for the treatment of lupus nephritis remain pathetically small19. Two quite distinct therapeutic problems emerge:
•The induction treatment of severe, acute life-threatening disease, often affecting many systems and usually near the onset of the disease; here the threat of the disease is paramount.
•There follows the maintenance treatment and long-term management of chronic, more or less indolent disease,
during which protection from the side-effects of treatment becomes more and more important.
The evidence for treating all but the mildest types of lupus nephritis with corticosteroids is very strong, but no prospective trial directly comparing a corticosteroid-treated group with one not so treated has ever been performed – nor is such a trial likely to be mounted.
Lupus Nephritis
In most patients with absent or trivial urine abnormalities, the biopsy appearances will be bland, the outlook good, and treatment unnecessary. Whether treatment with corticosteroids at this point might prevent subsequent evolution of severe disease has never been tested. There is equally little evidence that early treatment alters subsequent evolution of the disease in those with definite but minor urine abnormalities, normal renal function and mild histopathologic appearances (WHO class II). Nor is their clear evidence whether the outcome of membranous nephropathy (WHO class V) improves following treatment, although most clinicians do treat patients with this pattern at least with corticosteroids. Certainly, a proportion of such patients may slowly develop renal failure.
Therefore, it is in those groups with focal proliferative nephritis of varying severity (WHO class III), or severe diffuse proliferative nephritis (WHO class IV), that immunosuppression seems to have most to offer.
The acute phase: induction treatment
Corticosteroids
High-dose oral corticosteroids (starting dose: 60 mg/24 h) carry a heavy penalty in terms of side effects. Thus many clinicians believe a few (1–3) ‘pulse’ intravenous methylprednisolone infusions (0.5–1.0 g) combined with low-dose oral prednisolone (10–20 mg/day) can reduce the incidence of these side effects, particularly the altered facial appearance. Side effects following intravenous injection of high doses of corticosteroids are more common in children and adolescents, and include: cardiac arrhythmias or even cardiac arrest if given through central venous lines; unpleasant flushing sensations; acute hypertension; and very occasionally acute psychosis. However the only controlled trial comparing ‘pulse’ and oral corticosteroids was with small numbers of patients with mainly nonrenal lupus.
Cytotoxic agents |
|
Today, most physicians use cytotoxic agents in both the acute |
|
and maintenance phases of the treatment of severe lupus |
|
nephritis19,20, a strategy reinforced by results of recent meta- |
|
analyses (see below), although there are contrary views17,18,21. |
|
The choice of agent and the route of administration, however, |
|
remain controversial19. |
|
Cyclophosphamide has the advantage for the induction phase |
|
that it is a much more powerful inhibitor of B cells than is |
|
azathioprine, and the resynthesis of autoantibodies is reduced |
|
to normal levels rapidly and efficiently. Therefore most clini- |
|
cians prefer it for induction therapy. Additional benefit from |
|
intravenous rather than oral administration during the acute |
|
phase is unproven20,21 and undoubtedly carries a penalty in |
|
side effects in that leukopenia is deliberately induced. In spite |
|
of this, intravenous cyclophosphamide has become virtually |
|
the standard initial management for severe nephritis, although |
|
often using a regimen less aggressive than that carefully eval- |
|
uated by the NIH group (0.75–1 g/m2 monthly), and therefore |
|
of uncertain benefit. In addition it does not seem logical to use |
|
a drug intravenously that was specifically developed for oral |
|
use, which has to be extensively metabolized before it is active |
|
so that (unlike methylprednisolone) there is no immediate |
|
effect. Since the patient is usually in hospital at this point, |
|
compliance should not be an issue. |
365 |

Section 5
Glomerular Disease
Plasma exchange
Although there is an obvious rationale for plasma exchange in lupus, no role in acute severe lupus nephritis has been defined except perhaps in patient with vasculitis and/or cryoglobulinemia. Two controlled trials in severe lupus nephritis22 showed no benefit from the addition of thrice-weekly plasma exchange over conventional, combined cytotoxic and corticosteroid therapy, even when synchronized with plasma exchange to minimize antibody rebound. Other groups have used a more intensive course of exchange, one plasma volume daily for 7–10 days, but it is unknown if this extra treatment confers benefit.
Practical treatment guidelines for the acute phase of lupus nephritis are shown in Figure 27.9.
The chronic phase: maintenance treatment
Usually the acute disease will be under control after 12 weeks or less, although occasionally patients may have a stormy course and require several courses of ‘pulse’ methylprednisolone. The balance of benefit between strategies to avoid relapses or smoldering disease activity and the many side effects of the drugs are poorly evaluated, despite a number of controlled trials and a large amount of anecdotal information.
Corticosteroids
Corticosteroids remain the backbone of treatment in the maintenance as well as the acute phase; there are no studies of other treatments without prednisolone. To minimize the side effects of long-term corticosteroids, the dosage should be limited to prednisolone 5–15 mg/day. Daily and alternate-day regimens have not been formally compared in lupus. Monthly ‘pulses’ of methylprednisolone have been used also, but again the toxicity and benefits of this regimen have never been investigated.
Cytotoxic agents
Meta-analyses are unequivocally in favor of an additional clinical benefit of a cytotoxic agent during the maintenance phase when used in combination with corticosteroids23,24. Further, the long-term follow-up of the NIH trials have shown less progression of renal scarring at 10–15 years in those groups treated with a cytotoxic agent than in those treated with prednisolone alone25 (Fig. 27.10). However, in these studies it was not possible to distinguish any difference between outcomes of the (rather small) groups receiving the various cytotoxic drugs used (cyclophosphamide, azathioprine or both), or the route of administration. Nevertheless only a group treated with intravenous cyclophosphamide showed improved survival in contrast to a group of (partially historic) controls treated only with prednisolone26,27 but this difference in outcome did not become apparent until more than 5 years’ follow-up27.
The use of oral cyclophosphamide for longer than about 12 weeks should always be avoided because of bladder but also gonadal toxicity. Therefore, regular monthly, then bimonthly intravenous cyclophosphamide (0.75–1 g/m2) has been advocated for up to 2 years because the mediumand long-term results are superior to those obtainable with prednisolone
alone. However, it has become evident that the intravenous
366
Induction treatment for lupus nephritis
Mild nephritis |
Severe nephritis |
WHO class II, III (mild), V |
WHO class III (severe) + IV |
Proteinuria |
Nephrotic syndrome |
Normal renal function |
Reduced renal function |
|
Hypertension |
Prednisolone 10–15 mg daily |
|
If renal function depressed, add:
Azathioprine 2 mg/kg per day
Methylprednisolone i.v. I.0 g for 3 days (repeat if necessary)
Oral
Prednisolone 10–15 mg daily
and
Cyclophosphamide
1–3 mg/kg per day (reduced dose if renal impairment) after 12 weeks change to Azathioprine 2 mg/kg per day
If clinical vasculitis:
Plasma exchange 3–4 L daily for 7 days
Figure 27.9 Induction treatment for lupus nephritis.
cyclophosphamide regimen, like prolonged daily oral treatment, carries a considerable doseand age-dependent risk of gonadal damage with late menarche and early menopause being regular findings28. Relapse is common29 and the very long-term outcome in terms of stable remission still unclear. There is an increased risk of infection compared to corticosteroids alone.
Gonadal damage could in theory be limited by giving the pulse timed so that a developing follicle is not present, but pregnancy cannot be contemplated during such treatment, although it may be possible at a later date in those who do not become sterile. The oncogenic risk of such regimens may not be evident for many years and, apart from the bladder, this risk remains whatever route of administration is used. However, a major advantage of the intravenous regimen is that in noncompliant patients it permits low corticosteroid dosage with acceptable effects on appearance, and the treatment can be given under observation. A recent controlled trial showed that combined treatment with monthly pulses of both intravenous cyclophosphamide and methylprednisolone together carried long-term advantages over either treatment alone30.
Azathioprine in doses of 2–2.5 mg/kg/24 h has proved remarkably safe in the very long term20, although macrocytosis is very common and higher doses will induce leukopenia. Previously we used this agent during the acute phase of the disease but in the past decade we have used oral cyclophosphamide initially and then transferred patients to azathioprine after 12 weeks’ induction (see Fig. 27.9).
There is evidence that the addition of azathioprine does not increase infections; which mostly depend upon corticosteroid