

Лабораторная работа 1
.docxЛабораторная работа №1 «Неэмпирический квантово-химический расчет молекулы CFCl3»
Цели и задачи расчёта
Цель расчёта:
Определить критериев выбора и изучение принципов построения стандартного базисного набора для расчета молекулярных систем. Изучение методов интерпретации результатов расчета и представления молекулы в виде вектора свойств на их основе. Знакомство с программным комплексом GAMESS.
Задача расчета:
Выбрать наименьший из возможных оптимальный базис для неэмпирического расчета длин связей и валентных углов молекулы CFCl3 по программному комплексу GAMESS с точностью порядка 0.01 Å для длин связей и 1 градус для валентных углов, сравнимой с экспериментальной. На основании результатов расчета оценить стабильность и факторы, определяющие реакционную способность этой молекулы.
Характеристика и параметры расчета
Расчет молекулы CFCl3 осуществлен по программному комплексу GAMESS. Оптимизация геометрии проводилась методом Хартри-Фока в минимальном базисном наборе (RHF/STO-3G). Дополнительно, из оптимизированной ранее геометрии, были проведены несколько расчетов энергии методами RHF/6-31G* и MP2/6-31G*.
Интерпретация результатов расчёта
Оценка термодинамической стабильности молекулы
Энтальпия образования молекулы CFCl3 из простых веществ равна:
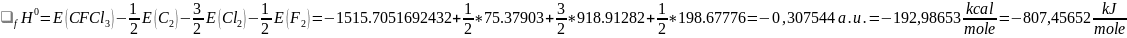
Вывод: Молекула CFCl3 стабильна при стандартных условиях.
Уточненная геометрия, порядки связей молекулы и валентность на атомах
Таблица 1. Равновесные геометрические параметры и порядки связей молекулы CFCl3
Связь |
Длина, Å |
Порядок связи |
Валентный угол |
град |
C – F |
1.360 |
0.857 |
F – C – Cl |
108.27 |
C – Cl |
1.811 |
0.989 |
|
|
Валентность атомов по Коулсону в молекуле CFCl3:
-
№
Атом
Валентность
1
F
0.896
2
C
3.825
3
Cl
1.008
Энергетическая диаграмма молекул
—
Оценка влияния базисного набора и метода расчета на величину полной энергии системы
Для оптимизации геометрических параметров был использован минимальный базисный набор STO-3G. Это минимально возможный базис для проведения любого квантово-химического расчета.
Чтобы учесть природу соединения и различные эффекты, которые могут влиять на полную энергию системы, были проведены дополнительные расчеты энергии различными методам. Результаты проведенных расчетов сведены в таблицу.
-
№
Метод/базис
Число базисных функций
E, а.е.
ΔEn-(n+1)
1
RHF/STO-3G
37
-1499.1773220350
—
2
RHF/6-31G*
87
-1515.7051692432
16.53
3
MP2/6-31G*
87
-1515.7051692432
0
В результате анализа данных в сравнительной таблице можно заметить, что расширение базисного набора влияет на результаты расчета, т.к. полная энергия системы с увеличением числа базисных функций понизилась. При расчете MP2/6-31G* по сравнению с RHF/6-31G* разница в энергии нулевой, что говорит о меньшем влиянии корреляции электронов в молекуле CFCl3 на полную энергию системы.
Анализ заселенности и распределение зарядов в исследуемом соединении по Малликену. Определение положения реакционных центров
-
Атом
Заселенность по Малликену
Заряд по Малликену
C
5.789389
0.210611
F
9.085177
-0.085177
Cl
17.041806
-0.041806
Максимальный отрицательный заряд концентрируется на атомах F.
Вывод: Атомы F – наиболее вероятные центры электрофильной атаки.
Оценка величины дипольного момента молекулы
-
DX
DY
DZ
/D/ (DEBYE)
0.057366
-0.223083
0.459215
0.513747
Электрический дипольный момент молекулы CFCl3 имеет небольшую величину 0.459 D.
Вывод: Молекула CFCl3 растворима преимущественно в слабо полярных растворителях.