

Villa
+
free
vWF
TFPI
XagBViia
Рис.
20.1.
Схема реакций, следующих за активацией
факторов X и IX
комплексом
TF-Vlia.
Она
отражает комбинированное влияние
факторов на конечное образование
тромбина. Необходимо отметить, что
малое количество тромбина, формируемое
комплексом TF-Vlla,
достаточно
для активации тромбоцитов, кофакторов,
а также фактора XI. Затем фактор Xla
способствует
большему образованию фактора IXa
из
фактора IX, усиливая
тем
самым гененерацию тромбина TFPI
—
ингибитор пути тканевого фактора; VWF
—
фактор Виллебранда; TF
—
тканевый фактор; Ха, Vila,
Xla и
пр. — активированные факторы свертывания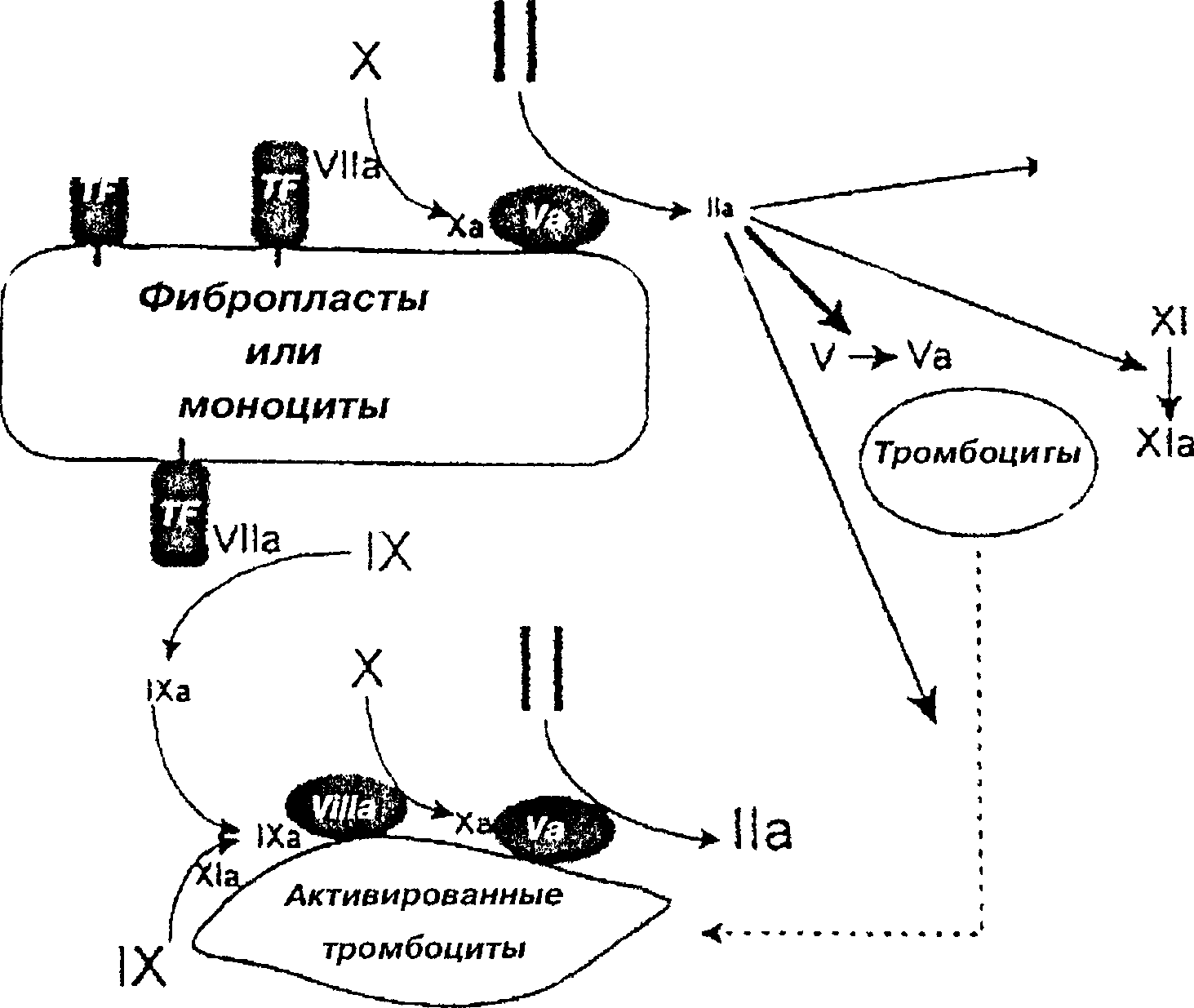
VIll/vWf I
Тромбоцитарный актомиозиноподобный белок ретрактозим обеспечивает консолидацию тромбоцитарно-фибринового сгустка и гемостаз.
Избыток образования фибрина ограничивается механизмом фиб- ринолиза, который протекает внутри самого сгустка, так как именно там создаются благоприятные условия для контакта плазминогена с его активатором при отсутствии его ингибиторов.
При этом, конечно, следует иметь в виду постоянное действие антикоагулянтов. И антитромбин, и протеины С и S, так же как и ингибитор пути тканевого фактора, работают постоянно и регулируют образование необходимого для организма количества фибрина (см. рис. 20.1).
Нарушения, возникающие при недостаточном образовании фибрина или при неполноценности функционирования тромбоцитарного звена гемостаза, так же как и при избыточной активности плазмина или неполноценности функционирования компонентов сосудистой стенки, приводят к нарушению процесса гемостаза в целом. Это и является основными механизмами развития феномена кровоточивости. Напротив, избыточное формирование фибрина вследствие неполноценности антикоагулянтов или слабости фибринолитического звена наряду с дефектами сосудистой стенки становится причиной избыточного внутрисосудистого свертывания крови, которое может приводить к нарушению проходимости сосудов или полной их окклюзии тромботическими массами — развитию феномена тромбоза с соответствующими последствиями. Иногда наблюдаются сочетания тромботических и геморрагических проявлений.
Таким образом, при нарушении функционирования системы гемокоагуляции различаются следующие синдромы: геморрагический, тром- ботический и тромбогеморрагический, синдром диссеминированного внутрисосудистого свертывания (ДВС-синдром).
20.2. Геморрагические синдромы
Геморрагический синдром — патологическое состояние, характеризующееся избыточной кровоточивостью.
Патогенетическая классификация геморрагий.
Дефект тромбоцитарного звена:
количественный — тромбоцитопении;
качественный — тромбоцитопатии;
смешанный.
Дефект прокоагулянтного звена — дефект фибринообразования:
количественный дефект факторов свертывания крови (недостаток факторов I, II, V, VII, VIII, IX, X, XI);
дефект структуры молекулы факторов свертывания крови;
наличие ингибиторов факторов свертывания.
Избыточная активность фибринолиза:
экзогенного происхождения;
эндогенного происхождения.
Дефект сосудистой стенки.
Комбинированный дефект компонентов гемостаза.
Геморрагические состояния, обусловленные дефектом тромбоцитарного звена. В норме количество тромбоцитов должно быть в пределах 2,0—4,0x1012/л.
Считают, что для обеспечения полноценного гемостаза достаточно количества тромбоцитов в пределах 30 ООО в 1 мм3. Некоторые авторы полагают, что даже меньшее количество полноценных тромбоцитов (15 ООО и даже 10 ООО в 1 мм3) способно обеспечить полноценный гемостаз. Геморрагическая тромбоцитопения была одной из первых идентифицированных геморрагических заболеваний человека. Ее описал Верльгоф еще в XVIII в. задолго дс того, как были обнаружены сами тромбоциты. В настоящее время различают несколько типов тромбоцитопе- ний (табл. 20.4).
|
Таблица 20.4 Классификация тромбоцитопений Тромбоцитопения | |||
|
вследствие нарушения образования |
вследствие повышенного разрушения |
вследствие перераспределения |
вследствие комбинации причин |
|
Сниженный мега- кариоцитопоэз: а) Наследуемые нарушения (гипо- пластическая тромбоцитопения с отсутствием радиальной кости — ТАР, мак- ротромбоцитопе- ния, синдром Фанкони и др.) б) внутриутробные изменения (краснуха, цитомегалови- рус и пр.), прием матерью тиазидов и пр.; |
Иммунные механизмы:
а) антитела пост- трансфузионные, антитела вследствие неонатальных конфликтов и пр.; б) антитела вследствие лекарственной аллергии: сульфаниламиды, хинины,препараты золота и пр.; в) следствие иных иммунных заболеваний: коллагено- зы, лимфопролиферативные болезни и пр.; г) инфекционные заболевания — бактериальные, вирусные (ВИЧ, Эпштейна—Барра и др.) и пр. |
Гиперспленоме- галия: застойная, инфильтративная, воспалительная, инфекционная и пр. |
Алкогольная болезнь печени |
|
Приобретенная гипоплазия: а) химические воздействия, в т.ч. лекарственные; б) инфекционные, в) аутоиммунные, г) алкогольные, д) иные (в т.ч. идиопатические) |
Потребление при заболеваниях: тромботическая тромбоцитемичес- кая пурпура, гемо- литико-уремичес- кий синдром, сердечно-сосудис- тые протезы, преждевременное |
Разведение крови при переливании старой крови |
Применение аппарата сердце—легкие |
|
Тромбоцитопения | |||
|
вследствие нарушения образования |
вследствие повышенного разрушения |
вследствие перераспределения |
вследствие коминации причин |
|
|
отслоение плаценты, эмболия околоплодными водами, внутриутробная гибель плода, токсикоз беременных и пр |
|
|
|
Неэффективный тромбоцитопоэз |
Тромбоцитопении при отдельных заболеваниях: болезнь Виллебранда |
Гипотермия |
Лимфопроли- феративные заболевания |
|
а) Наследуемые: аутосомальная доминантная, аномалия Мая— Хегглина, синдром Вискотта— Олдриджа и др. б) Дефицит витамина В12, фолиевой кислоты и др. в) Прочие; паро- ксизмальная ночная гемоглобин- урия, синдром ди Гульельмо и пр. |
Смешанные механизмы |
|
Прочие |
Тромбоцитопатии — тромбоцитарные дисфункции. Нарушение в тромбоцитарном звене могут приводить к кровоточивости не только при количественном недостатке тромбоцитов, но и при их неполноценности. Неполноценность тромбоцитарного звена характеризуется неспособностью тромбоцитов осуществлять феномены адгезии, агрегации, высвобождения внутреннего содержимого, содержащегося в гранулах, влиять на взаимодействие факторов коагуляции на фосфолипидной поверхности, и неполноценностью феномена ретракции. Эти нарушения тромбоцитарного звена получили название тромбоцитопатии. Первая тромбоци- топатия была описана Эдвардом Гланцманом еще в 1920 г., и ее называли болезнью Гланцмана. Известно довольно большое количество тромбоцитопатии. Существует несколько подходов к их классифицированию. Например, предлагается характеризовать нарушения отдельных тромбо- цитарных функций.
Тромбоцитарные дисфункции включают следующие формы расстройства:
Нарушения адгезии:
дефект гликопротеида 1Ь — болезнь Бернара-Сулье (в сочетании с умеренной тромбоцитопенией и гигантскими тромбоцитами);
псевдоболезнь фон Виллебранда (отсутствие реакции на не- мультимерные белки Виллебранда) и др.;
Нарушения агрегации:
болезнь Гланцмана — нарушение агрегации с АДФ и коллагеном вследствие дефекта комплекса гликопротеинов lib—Ша;
дефект агрегации в ответ на коллаген (нарушение гликопротеида la);
дефект аггрегации в ответ на тромбин, дефект агрегации вследствие нарушений коллагена (тип IV, III —синдромы Эй- лерса—Данлоса, Марфана, несовершенного остеогенеза).
Нарушения прокоагулянтной активности тромбоцитов:
первичный прокоагулянтный дефект тромбоцитарной мембраны в связывании фактора V при нормальной агрегации, секреции и нормальном времени кровотечения;
Нарушения реакции высвобождения:
дефект плотных телец (нарушение высвобождения адеииннук- леотидов или серотонина, или кислых гидролаз, или арахидоновой кислоты под действием АДФ, коллагена и адреналина);
синдром серыхтромбоцитов — (отсутствие а-гранул, содержащих фибриноген, фактор Виллебранда, тромбоспондин и др.)
Нарушения ретракции:
тромбастения Гланцмана;
Сочетанные нарушения функций:
синдром Херманского—Пудлака — дефект высвобождения из плотных телец и из а-гранул в сочетании с окулокутанным альбинизмом и наличием пигментных .макрофагов в костном мозге;
синдром Вискотта—Олдриджа — тромбоцитопения, тромбоци- топатия (дефект высвобождения из плотных тел, дефект агрегации на адреналин) в сочетании с экземой и иммунодефицитом;
аномалия Мэя—Хегглина — гигантские тромбоциты, тромбоцитопения, аномальные гранулоциты с крупными включениями;
синдром Чедиака—Хигаши — тромбоцитопатия (дефект плотных телец) в сочетании с тромбоцитопенией и дефектом пигментации (волосы, кожа, сетчатка);
TAR-синдром — тромбоцитопения при отсутствии радиальной кости и тромбоцитопатией (дефект высвобождения из плотных телец).
Кроме перечисленных наследуемых заболеваний, кровоточивость нередко вызывается механизмами, связанными с приобретенными дефектами функций тромбоцитов, которые чаще всего наблюдаются при приеме лекарств (ацетилсалициловая кислота и др.), уремии, операциях с применением аппарата «сердце—легкие» и др.
Следует отметить, что изолированные дефекты тромбоцитарного звена независимо от патогенетического механизма клинически проявляются сходно. Для них характерны «синяки», петехии, экхимозы, кровотечения при повреждениях кожи и слизистых оболочек, маточные, носовые и кишечные кровотечения.
Геморрагии при нарушениях образования фибрина. Дефект прокоагулянтного звена. Дефект каждого из факторов прокоагулянтного ззена может возникать вследствие недостаточной их продукции, но также при образовании антител к факторам свертывания, а иногда вследствие формирования дефектной молекулярной структуры.
Описаны геморрагические состояния при недостаточности следующих факторов свертывания крови: 1, II, V, VII, VIII, IX, X, XI, XIII. Механизм геморрагических состояний при дефекте фактора XII, прекалликреина, ки- ниногена высокой молекулярной массы, Са2+ и тканевого фактора точно не известен. В тех же случаях, когда кровоточивость предполагается именно их недостаточностью, она настолько незначительна, что не требует коррекции. Наиболее часто приходится встречаться с геморрагическими явлениями вследствие дефицита фактора VIII, который бывает у одного из 4000—20 ООО человек. В 5—10 раз реже отмечается другая патология, обусловленная дефектом фактора IX, — гемофилия. Гемофилия А развивается при дефекте фактора VIII, гемофилия В — при дефекте фактора IX. Проявления гемофилии характеризуются кровоточивостью в суставы, образованием гематом в мышцах, кровоизлияними в полость черепа. Возможны случаи кровотечений при укусах языка, повреждении его уздечки.
При гемофилиях крайне редко возникают петехиальные кровотечения, кожная пурпура, кровотечения из слизистых оболочек, нередки гематурии. Геморрагические диатезы вследствие дефицита факторов II, V, VII, X и XI встречаются значительно реже. Проявления этих расстройств сходны с таковыми у больных с дефицитами факторов VIII и IX, хотя их интенсивность выражена значительно слабее. В связи с этим высказывается предложение называть любое из дефектов фибринообразования гемофилией, что не лишено смысла.
Геморрагический синдром при избыточной фибринолитичес- кой активности. Часто причиной этих расстройств является усиление фибринолиза, вызванное экзогенными факторами, нередко при проведении тромболитической терапии. Описаны случаи гиперфибринолити- ческих кровотечений, обусловленных недостаточным контролем за активным плазмином со стороны а2-антиплазмина. Последний может быть как сниженным, так и неполноценным. Гиперфибринолиз часто отмечается в комбинации с избыточным свертыванием крови. Проявления гиперфиб- ринолиза идентичны таковым при дефектах формирования фибрина.
Геморрагические состояния вследствие дефекта сосудистой стенки. Причиной геморрагического синдрома может быть неполноценность сосудистой стенки, определяющая ее неспособность осуществлять эффективный гемостаз без нарушений ее целости. Этот механизм обусловливает развитие патологии, относящейся к геморрагическим телеан- гиэктазиям; она проявляется формированием неполноценных сосудов, стенка которых в отдельных местах состоит лишь из одних эндотелиальных клеток, лишенных мышечного слоя и даже эластической мембраны. Эти участки значительно чаще подвержены разрывам, чем нормальные сосуды, и сопровождаются кровотечениями.
Аномалия коллагена рассматривается в качестве основного механизма, определяющего неполноценность сосудистой стенки и гемостаза при генерализованной фибродисплазии эластических волокон, при дегенеративных повреждениях соединительной ткани, сочетающейся с гипоплазией эластических волокон и аномалией сосудов. Хорошо известны сосудистые геморрагии при дефиците витамина С. Приобретенные дефекты сосудистой стенки проявляются геморрагиями при аутоиммунных заболеваниях— анафилактоидной пурпуре (болезнь Шенлейна—Геноха), васку- литах, а также при изменении сосудистой стенки вследствие отложения в ней аномальных белков (миеломная болезнь, макроглобулинемия и пр.).
Геморрагические синдромы, вызываемые сочетанием нескольких дефектов гемокоагуляции. Наиболее часто такие геморрагии наблюдаются при тромбогеморрагическом синдроме, называемом также синдромом диссеминированного внутрисосудистого свертывания крови (ДВС-синдром) или же коагулопатией потребления. При этом у пациентов отмечается резкое снижение в плазме как факторов свертывания крови, так и уровня тромбоцитов. Одновременно у многих из них имеет место выраженный гиперфибринолиз. К этой же группе необходимо относить наиболее часто встречающуюся геморрагию — болезнь Виллебранда. В основе ее лежит дефицит специфического протеина, называемого белком Виллебранда, который действует двояко: с одной стороны, он обеспечивает контакт тромбоцитов с субэндотелиальными структурами за счет взаимодействия с тромбоцитарным гликопротеидом lb и субэндотелием, а с другой стороны, белок Виллебранда обеспечивает стабильность антигемофилического фактора VIII. Кровоточивость при этом заболевании имеет сходство с кровоточивостью при дефекте образования фибрина, а также с кровоточивостью, характерной для дефектов тром- боцитарного звена.
20-3. Патология, обусловленная гиперкоагуляцией: тромботический синдром
Тромботический синдром, или синдромы, — патологические состояния, характеризующиеся внутрисосудистым или внутрисердечным формированием кровяных сгустков — тромбов, образующихся за счет фибрина, тромбоцитов и оседающих в их структурах тех или иных количеств эритроцитов и лейкоцитов, связанных со стенкой сосудов и в той или иной степени обтурирующих их просвет. Какова причина тромбозов? Еще в середине прошлого века Рудольф Вирхов определил свою знаменитую «тромботическую триаду», согласно которой причинами тромбозов могут быть следующие факторы: нарушение целости сосудистой стенки, замедление кровотока и повышенная способность крови к свертыванию. К настоящему времени известно большое число факторов, с помощью которых стенка сосуда способна влиять на внутрисосудистые тромбооб- разования: на степень спазмирования — через эндотелиальный фактор релаксации — оксид азота, эндотелин-1, ангиотензинпревращающий фактор, за счет влияния на тромбоциты — через синтезируемый эндотелием фактор Виллебранда, фактор, активирующий тромбоциты, проста- циклин, влияющий на функцию тромбоцитов белок теплового шока, Р- селектин и др. На поверхности эндотелия происходит активация факторов V, VIII, X, возникающая вследствие секреции эндотелиальными клетками ингибитора тканевого фактора и продукции протеина S, синтеза тканевого активатора плазминогена и урокиназы, а также ингибиторов активаторов плазминогена РАМ, PAI-2.
Степень обтурации просвета сосуда определяет тяжесть расстройств. Полная обтурация артериального сосуда вызывает ишемию той части органа, кровоснабжение которой она определяет. При быстрой обтурации обычно развивается инфаркт, т.е. омертвение той или иной части органа, которая была полностью лишена доступа крови. При медленной обтурации сосуда возможно развитие коллатерального кровообращения, которое в какой-то степени может ограничить размеры и степень ишемии тканей органа. При развитии тромбов в венах последствия менее драматичны, так как венозная система обычно имеет множество параллельных путей венозного кровотока. Однако тяжесть также определяется размерами окклюзированного венозного сосуда. Чем больше эти размеры, тем труднее развиться коллатеральному кровотоку, адекватному объему крови, оттекающей от региона, принадлежащего ктромбируемой вене.
Венозный тромб определяет не только замедление кровотока, но и степень сдавления окружающих тканей застойной кровью, в т.ч. и артерий, находящихся в зоне поражения. Для венозных тромбов в большей степени, чем для артериальных, характерна способность его частиц отрываться от основной массы тромба, превращаться в эмболы и разноситься током крови в сторону сердца и далее — от него. Но и артериальные, и внутрисердечные тромбы способны вызвать эмболию, которая может определять характер расстройств зависимый от места и объема сосуда.
Известно, что основной причиной артериальных тромбов является атеросклероз сосудов. Нарушение целостности атеросклеротических бляшек является наиболее драматическим процессом их эволюции, который недавно получил название атеротромбоза. При их разрыве высвобождающийся тканевый фактор включает каскад фибринообразования, который наряду с активизацией тромбоцитарного компонента приводит к образованию тромба на месте повреждения. Имеется предположение, что в основе формирования атеросклеротических бляшек большая роль также принадлежит внутрисосудистому свертыванию крови.
Установлено, что при сердечной недостаточности и замедлении кровотока при уменьшении активности мышц, особенно икроножных, частота венозных тромбозов резко увеличивается. И наоборот — ускорение венозного кровотока при стимуляции мышц, в т.ч. искусственной, повышении тонуса венозных сосудов и при уменьшении степени сердечной недостаточности частота венозных тромбозов уменьшается.
Большая роль в формировании тромбов, больше венозных, но также и артериальных, отводится врожденной предрасположенности к их формированию — наличиютакназываемойтромбофилии. Начало учению о тромбофилиях было положено лишь в 1965 году, когда норвежскому ученому О. Эгебергу удалось доказать, что в одной из норвежских семей частые тромбозы связаны с выраженным снижением в их организме антитромбина III, естественного противотромботического фактора человека. Дальнейшие исследования привели к тому, что на сегодня имеется целый перечень состояний, при которых образование тромбов сопровождается дефектом тех или иных противотромботических приспособлений организма.
Тромбофилические состояния включают следующие нарушения:
дефект антитромбина III:
количественный;
качественный, смешанный;
дефект протеина С;
дефект протеина S;
резистентность к активированному протеину С-дефект фактора V (Лейдена);
дефект молекулы протромбина — протромбин 20210;
дефект фибриногена;
дефект кофактора гепарина II;
дефект плазминогена;
дефект активатора плазминогена;
дефект фактора Хагемана;
наследуемая гиперактивность тромбоцитов;
гипергомоцистеинемия;
люпус-антикоагулянт.
Все перечисленные состояния характеризуются повышенной возможностью организма к развитию внутрисосудистых тромбов, однако их наличие не является гарантией обязательного тромбообразования. По- видимому, для этого нужны какие-то добавочные компоненты — повреждение сосудистой стенки или замедление кровотока. Вероятно, что при образовании тромба всегда имеет место сочетание нескольких предрасполагающих факторов.