

4.3. Иммунодефицитные состояния
Как и любые системы организма, иммунная система подвержена патологическим процессам. Выделяют четыре основных типа иммунопатологии:
иммунная недостаточность (иммунодефициты) вследствие дефектов развития или действия повреждающих факторов;
гиперчувствительность, или измененная реактивность, основной формой которой является аллергия;
аутоиммунная патология;
опухоли иммунной системы, прежде всего лимфопролиферативные процессы.
Ниже будут кратко охарактеризованы иммунодефициты, аутоиммунные и лимфопролиферативные процессы.
Иммунодефициты разделяют на две группы — первичные (врожденные), как правило, имеющие наследственную природу, и вторичные (приобретенные), вызванные различными воздействиями, как эндогенными (болезни), так и экзогенными (действием агрессивных физических и химических факторов). Наиболее характерным клиническим проявлением иммунодефицитов служит высокая подверженность заболеваниям, вызываемым микроорганизмами, в частности простудным; для иммуноде- фицитных состояний характерна связь инфекционных заболеваний с оппортунистическими агентами (т.е. сапрофитами, в норме непатогенными). При некоторых формах иммунодефицитов повышается риск развития опухолей.
4.3.1. Первичные иммунодефициты
Гоуппу первичных иммунодефицитов образуют заболевания, в основе которых лежит наследственно обусловленная дефектность структуры и функционирования иммунной системы, которая проявляется в нарушении иммунной защиты.
Первичные иммунодефициты — это очень редкие состояния (примерно 1 больной на 1 ООО ООО человек). Они являются почти исключительно уделом детского возраста, поскольку значительная часть больных с тяжелыми формами иммунодефицитов не доживает до 20 лет, а при более легких формах иммунологические дефекты с возрастом в определенной степени компенсируются.
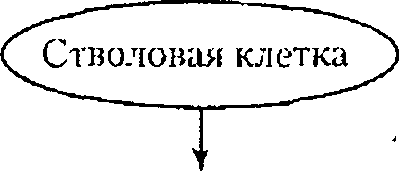
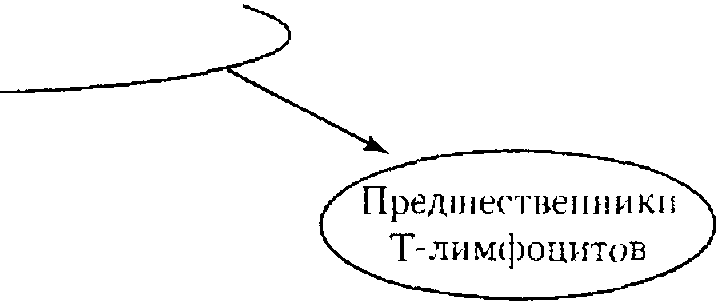
Как правило, в основе первичных иммунодефицитов лежит генетически обусловленный блок развития клеток иммунной системы или выпадение важных иммунных процессов вследствие дефекта определенных молекул, например ферментов или мембранных структур (схема 4.3).

Общий
лимфоидаын предшественник
в
Тяжелый комбинированный иммунодефициты (варианты, связанные с дефектами перестройки рецепторных-генов)
Х-сцеплепная
агамма- глобулинемия (дефеки тирозиНкиназы
btk
Синдром
ди Джоржи Синдром Незелова (дисгенезии
тимус)
Дефицит CD3 клеток (дефециты генов у и q пеней)
Синдром Вискота-Оддрича (дефект CD43)
Атаксация-телеангиоэктазия (дефекты репарации ДНК
)
Тяжелый комбинированный иммупнодефецит (варианты, связанные е дефектами АДА и ПНФ)
![]()
Зрелые
В-КЛС1КИ![]()
![]()
Зрелые Т-клетки
)
|
Нарушения формирования, субпопуляций | |
|
Дефект развития СБ4+клеток (дефекты генов PEXnMIIC II) |
Дефект развития СОВ^клеток (дефект ZAP-70) |
|
| |
|
Нарушение функционирования CD4+ | |
|
rmiep-IgM-синдром (нарушение переключения изотипои дефекта CD40L) | |

комбинированные иммунодефициты;
иммунодефициты с преимущественным поражением клеточного
иммунитета;
преимущественно гуморальные иммунодефициты.
К первым относят заболевания, в основе которых лежат генетические дефекты, затрагивающие различные линии дифференцировки лимфоцитов, а также ранние этапы их развития, общие для Т- и В-линий. Во вторую группу входят иммунодефициты, при которых нарушается развитие Т-клеток и страдают опосредуемые ими реакции клеточного иммунитета; к этой же группе относятся дефекты фагоцитирующих клеток. В группу гуморальных иммунодефицитов включают патологию, в основе которой лежит нарушение развития В-клеток и Т-хелперов гуморального ответа, а также патологию компонентов комплемента.
В последние годы выясняются молекулярные основы поражения при первичных иммунодефицитах. Одной из первых была расшифрована природа комбинированных иммунодефицитов, связанных с недостаточностью ферментов пуринового метаболизма. Известны варианты таких дефектов, обусловленные мутациями генов, кодирующих аденозиндеза- миназу и пуриннуклеотидфосфорилазу. Основой другой формы тяжелого комбинированного иммунодефицита, затрагивающего Т- и В-ростки лимфопоэза, служит дефект процесса перестройки генов антигенраспоз- нающих рецепторов, связанный с отсутствием ферментов рекомбиназ, которые катализируют этот процесс.
Очень разнообразен спектр генетически обусловленных нарушений выработки антител. Их причиной может быть как поражение В-лимфоцитов (их развития или экспрессии генов иммуноглобулинов), так и дефектность Т-клеток (ослабление хелперной активности). Примером первого рода может служить агаммаглобулинемия Брутона, сцепленная с Х-хро- мосомой. Ее основой являются мутации гена, детерминирующего фермент тирозинкиназу btk, которая связана с антигенраспознающим рецептором В-лимфоцитов. Отсутствие этой тирозинкиназы делает невозможным развитие В-лимфоцитов уже на самых ранних стадиях.
В основе другого первичного иммунодефицита — гипер-1дМ-синд- рома лежит дефект CD154 — молекулы, появляющейся на поверхности Т-клеток при их активации; в результате ее взаимодействия с молекулой CD40 поверхности В-лимфоцитов в эти клетки передается сигнал, обеспечивающий их дифференцировку в антителообразующие клетки, а также переключение изотипов секретируемых антител. В отсутствие этого сигнала происходит синтез иммуноглобулинов только одного изотипа — IgM, что сопровождается ослаблением гуморального иммунного ответа. Существуют формы гуморальных иммунодефицитов, при которых нарушено образование иммуноглобулинов какого-либо одного изотипа. Среди таких селективных дефектов наиболее частым является дефицит IgA.При нем присутствуют В-лимфоциты, несущие мембранный IgA, однако не образуются плазматические клетки, секретирующие 1дА-антитела.
Ряд комбинированных иммунодефицитов возникает при локализованных дефектах генов мембранных молекул адгезии. Следствием таких мутаций является нарушение миграции клеток, в первую очередь нейт- рофилов и моноцитов/макрофагов, а также их взаимодействий с клетками других типов. Примером могут служить сходные поражения, развивающиеся как результат наследственных дефектов экспрессии (Р2-интегри- нов и углеводных детерминант, распознаваемых селектином L. Эти поражения представляют собой два варианта LAD-синдрома — дефицит адгезии лейкоцитов, признаком которого является ослабление функции нейтрофилов, и повышение чувствительности к гнойным инфекциям.
Дефекты компонентов комплемента представлены вариантами с поражением практически всех основных факторов классического и альтернативного путей активации комплемента. Как правило, выпадение единичных компонентов системы комплемента проявляется в умеренном снижении устойчивости к некоторым возбудителям. Лишь дефицит ингибитора С1 q сопровождается развитием ангионевротического отека, обусловленного накоплением вазоактивных пептидов С5а и СЗа.
Иммунодефициты, в основе которых лежит дефект генов цитокинов, немногочисленны, что связано с «избыточностью» системы цитокинов, которая обусловлена взаимозаменяемостью их функций. Лишь когда генетический дефект затрагивает функцию многих цитокинов, это проявляется в тяжелых расстройствах иммунитета, что происходит, например, при дефекте гена у-цепи, общей для рецепторов интерлейкинов 2, 4, 7, 13 и 15.
В результате дефекта, затрагивающего ген мембранного сиалопро- теина CD43, развивается синдром Вискотта—Олдрича, о чем свидетельствует тромбоцитопения с геморрагическим синдромом в сочетании с экземой и комбинированным иммунодефицитом. При этом заболевании аномально функционирует цитоскелет, что отражается на подвижности клеток и межклеточных взаимодействиях, важных для осуществления иммунных процессов.
При атаксии-телеангиэктазии наблюдается поражение различных функций, обусловленное слабостью аппарата репарации ДНК и нестабильностью хромосом, а также дефектами клеточного цикла. Это дает неожиданное сочетание симптомов: комбинированный иммунодефицит (недоразвитие вилочковой железы, дефицит Т-клеток и иммуноглобулинов «поздних» изотипов — lgG2, lgG4, IgE, IgA), неврологические отклонения (атаксия), поражение сосудистой стенки (телеангиэктазии), нарушение пигментации.
Помимо рассмотренных «точечных» поражений иммунной системы известны первичные иммунодефициты, развитие которых обусловлено множественными дефектами, затрагивающими формирование в эмбриогенезе различных органов, включая органы иммунной системы. Так, наследственный порок, приводящий к нарушению развития у эмбрионов ч-1- ловека производных 3 и 4 жаберных щелей, служит основой синдрома Ди Джорджи с дефектом развития вилочковой железы (она не заселяется предшественниками Т-клеток, развитие которых прерывается на костномозговой стадии) и гистогенетически родственных органов (паращито- видных желез и т.д).
Основным симптомокомплексом, отражающим нарушение иммунной защиты при первичных иммунодефицитах, является инфекционный синдром, т.е. понижение резистентности к инфекционным агентам, в том числе сапрофитным (Pneumocystis carinii, Candida, цитомегаловирус, некоторые энтеровирусы). Характер нарушений иммунной защиты определяется локализацией поражения в иммунной системе. Так, при блокаде процесса перестройки рецепторных генов отсутствуют как Т-, так и В-клетки и не развиваются ни клеточные, ни гуморальные формы иммунного ответа. При селективных дефектах определенных классов лимфоцитов, а также их субпопуляций выпадают именно те иммунологические функции, за которые ответственны поражаемые типы клеток. При блокаде развития В-клеток развивается агаммаглобулинемия с нарушением гуморальной з&щиты от внеклеточных бактерий и их токсинов, а при дефицитах Т-лимфоцитов страдает клеточная защита от вирусов и микобактерий. При некоторых формах первичных иммунодефицитов (атаксия-телеангиэкста- зия, синдром Вискотта-Олдрича и т.д.) значительно повышается риск развития злокачественных опухолей (до 10—15%). Нередко нарушения иммунологических функций регистрируются при нормальной численности соответствующих клеток.
Клинико-иммунологическое обследование дает четкие результаты лишь при тех формах первичных иммунодефицитов, при которых точно локализован дефект. Так, при тяжелом комбинированном иммунодефиците отсутствуют как Т-, так и В-клетки, при синдроме Ди Джорджи резко снижено содержание Т-лимфоцитов, а при агаммаглобулинемиях — В-лимфоцитов. По изменению концентрации иммуноглобулинов в сыворотке или компонентов комплемента различных изотипов может быть установлена локализация дефекта в системе гуморального иммунитета. Все большую диагностическую значимость приобретает определение конкретных мембранных маркеров клеток иммунной системы (молекул адгезии, CD154, CD43 и т.д.), а также методы, позволяющие выявить мутации конкретных генов.