

5 курс / Пульмонология и фтизиатрия / Чучалин_А_Г_Респираторная_медицина_т_1_2017
.pdfРаздел 1
ческие экспрессионные векторы), в которых они затем размножаются [50]. Типичный репрезентативный космидный или фаговый банк человека содержит около 1 млн индивидуальных клонов, причем каждый — с маленькой собственной частью генома (от 20 000 до 40 000 базовых пар на клон). Если известны биологические функции или антигенные свойства связанного с заболеванием протеина, можно обследовать экспрессионные банки генов с помощью функциональных наборов или специфических антител (АТ) к клону, содержащему искомый ген. Если такой клон идентифицирован, он может быть размножен in vitro, при этом он производит огромное число копий гена, достаточное для характеристики гена до полного секвенирования и выделения продуктов протеина в чистой форме. Клонирование генов описанными методами — очень сложный процесс, особенно когда мало предварительной информации об искомом гене.
Полимеразная цепная реакция (ПЦР) и характеристика генных дефектов. Точным, хотя и дорогостоящим, методом для характеристики генов заболевания у индивидуальных пациентов можно считать клонирование этих генов и полное секвенирование. Грубую информацию о структурных генных повреждениях получают посредством так называемого рестрикционного анализа. При этом геномную ДНК пациентов разрезают с помощью специфических энзимов (рестрикционных эндонуклеаз), разделяют с помощью электрофореза и гибридизируют с помощью специфического генного зонда [48]. Из полученного таким образом рестрикционного образца распознаются большие генные дефекты, а часто даже патогенные точечные мутации. Современная технология, которую можно использовать для быстрого доказательства таких мутаций, и есть так называемая ПЦР [14]. Технология ПЦР используется для диагностики моногенных, а также инфекционных и злокачественных заболеваний. Ее принцип — размножение (амплификация) in vitro сегмента ДНК, имеющегося в мизерном количестве. Это автоматизированный процесс с использованием последовательно специфического праймера — олигонуклеотида и термоустойчивой синтезированной ДНК-полимеразы для энзиматического увеличения количества ДНК на выходе. На практике из нескольких микролитров крови пациента выделяют геномную лейкоцитарную ДНК, которая содержит мизерное количество необходимой для исследования целевой последовательности. Для структурного анализа (например, секвенирования) это количество было бы слишком незначительно, однако с помощью ПЦР оно амплифицируется более чем в 106 раз. Помимо геномной ДНК, могут амплифицироваться и секвенироваться индивидуальные РНК. Для этого сначала выделяют клеточную общую РНК, которая используется для синтеза in vitro комплементарной ДНК (сДНК),
азатем амплифицируется как обычная ДНК с помощью ПЦР.
Впоследнее время появились методы, позволяющие прямо секвенировать ПЦР-продукты из геномной ДНК без предшествующего клонирования [44]. Эта технология освобождает от необходимости клонирования индивидуального гена и имеет большое значение для характеристики генных дефектов. В распоряжении исследователя для реализации этой технологии имеются специальные радиоактивные и автоматизированные протоколы.
Генное сканирование. Хотя ПЦР-амплификация и прямое секвенирование — очень эффективные и быстрые методы для характеристики генных дефектов человека, все-таки пока есть определенные проблемы. Одна из них — обследование (сканирование) гена в целом на наличие точечных мутаций. Эта проблема возникает, например, при анализе кандидатных генов и подозрении, что они несут при определенном заболевании патогенные мутации, распределенные на протяжении всего гена. В качестве предварительного этапа секвенирования при такой проблеме разработали так называемую технику генного сканирования, которая позволяет ответить на вопрос, имеются ли вообще какие-либо мутации в обследуемой области. Тогда при наличии этой предварительной информации,
азатем при окончательной характеристике посредством секвенирования возможно ограничение на действительно нужной (позитивной) области обследования. Все техники сканирования базируются на ПЦР-технологии и при относительно небольшой трудоемкости позволяют обследовать гены на наличие мутаций у большого количества пробандов.
Трансфер генов in vivo. Существуют следующие принципы генной терапии.
•Дефектный ген, являющийся причиной моногенного наследственного заболевания, замещается на функционально способный ген (генная аугментация). Примером такой генной терапии может служить тяжелый комбинированный иммунодефицит, когда производится трансфер гена аденозиндезаминазы.
•Трансферируется ген, дополнительная экспрессия которого у пациентов при заболевании, не обязательно обусловленном дефектом гена, требует терапевтического воздействия. Примером этого может служить локальная экспрессия цитотоксической субстанции при малигноме.
•Дефектный ген замещается функционально способным (здоровым) геном (генная коррекция посредством гомологичной рекомбинации) [11].
Впервых клинических исследованиях по генной терапии генный трансфер производился ex vivo. Так, у пациента забирали целевые клетки генной терапии, например гепатоциты, клетки костного мозга или лимфоциты, изменяли их ген-
40

Анатомия и онтогенез респираторной системы
но-техническим способом вне организма, а затем вводили вновь. В последнее время разработаны такие векторы трансферирующих генов, которые позволяют проводить у людей прямой трансфер генов in vivo. Например, аденовирусные векторы применяют при генной терапии для введения нормального муковисцидозного CFTR-гена в эпителий дыхательных путей пациентов, страдающих МВ [10]. Сейчас интенсивно работают над развитием таких векторов, которые позволяют проводить органоспецифический in vivo трансфер в печень, костный мозг, эндотелий сосудов. Разрабатываются и другие способы соматической терапии коррекции гена.
Генное прицеливание (Gene Targeting). Для изучения сложных генетических патомеханизмов важно производство трансгенных животных посредством целевого изменения их генома. Животным создают генетические дефекты
ианализируют их биологическое влияние на организм в целом. Речь идет о функции таких мутированных генов, воздействие на организм которых неизвестно или известно лишь частично. У трансгенных животных определенные гены могут полностью инактивироваться (Gene Disruption) или целевым образом мутагенизироваться (Targeted Mutagenesis). Была создана модель человеческого МВ на мыши, для чего мутагенизировали мышиный гомолог гена человеческого МВ [7]. Близким способом была создана на мыши модель ААТ-дефицита человека. Создают
идругих трансгенных животных, например, для изучения энзима супероксиддисмутазы.
Известные гены риска респираторных заболеваний
Ген α-1-антитрипсина
В начале 1960-х годов из человеческой сыворотки был выделен трансингибиторный протеин, названный ААТ. ААТ — это гликопротеин острой фазы, он подавляет как трипсин, так и целый ряд серинпротеиназ, а его главная физиологическая роль заключается в ингибировании протеолитического энзима — нейтрофильной эластазы [32]. Дальнейшее изучение ААТ выявило связь между его дефицитом и хронической обструктивной болезнью легких (ХОБЛ). Далее были изучены структура, функции и клиническое значение этого протеиназного ингибитора (PI) и показано его ау- тосомно-рецессивное наследование. Дальнейшее наблюдение за пациентами с ХОБЛ, ассоциированных с ААТ, выявило, что курение значительно ухудшало их состояние и укорачивало жизнь почти на 20 лет [25]. Развитие способов изучения протеина позволило обнаружить новое число вариантов этого протеина, например дефектную аллель Z. Было показано, что некоторые ААТдефектные аллели связаны не только с ХОБЛ, но и
с заболеваниями печени: например, гомозиготный носитель Z-аллели был связан с циррозом печени у маленьких детей, тяжелой гепатопатии — с дефектом аллели Mmalton и Mduarte [9].
Дальнейший поиск причины связи между заболеваниями легких и недостатком ААТ привел к развитию так называемой протеиназно-антипро- теиназной теории. В огромном количестве исследований было показано, что различные протеолитические энзимы нарушают матричную структуру легкого, что ведет к неадекватной работе легких. ААТ — доминирующий протеиназный ингибитор в бронхоальвеолярном смыве (БАС). С недавних пор в распоряжении врачей имеются препараты ААТ, которые реконструируют в легком эластаз- но-ингибиторный потенциал сыворотки у больных с дефицитом ААТ. Его применяют внутривенно или в виде аэрозоля [22].
Развитие современных технологий открыло новые возможности для анализа ААТассоциированных заболеваний. В 1980-х годах ААТ-ген был локализован, клонирован и секвенирован на хромосоме 14 [42]. Затем были клонированы и секвенированы многочисленные дефекты ААТ-гена, охарактеризованы возможные мутации и их влияние на синтез и функцию ААТ. Клонирование и структурный анализ ААТ-гена позволили провести генетический синтез гликозилированного человеческого ААТ в фибробластах. Появилась возможность прямого трансфера и локальной экспрессии ААТ-гена в дыхательных путях in vivo с помощью аденовирусного вектора. Генный продукт ААТ уже можно использовать для внутривенного и ингаляционного введения.
Ген муковисцидоза
Кроме ААТ-гена, известен другой ген риска — причина МВ. В классических случаях эта болезнь начинается в раннем детстве с рецидивирующей легочной инфекции с выраженной бронхиальной дискринией и гиперпродукцией бронхиального секрета с последующим формированием бронхоэктазов (БЭ), рецидивирующими пневмониями и хронической обструкцией дыхательных путей. Больные умирают, как правило, рано. МВ часто ассоциируется с экзокринной панкреатической недостаточностью, следствием чего и является дискриния с развитием густого секрета. Причины этого сочетания до конца не выяснены. Помимо бронхов и поджелудочной железы, отмечается специфический дефект потовых желез: такие больные имеют необычно высокое содержание хлорида натрия в поте, что при сильной жаре ведет к его большим потерям, вплоть до развития коллапса.
МВ наряду с недостаточностью ААТ — самое частое наследственное заболевание белого населения Европы и Северной Америки, приводящее к смерти. При анализе сцепления CF-семей с помощью молекулярно-генетического маркера в 1985 г.
41

Раздел 1
установили локализацию CF-гена на хромосоме 7q 31–32 [52]. Более точную локализацию и клонирование провели в 1989 г. Название «CF-ген» следует из его обозначения — Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), этот ген частично секвенирован и клонирован на искусственной дрожжевой хромосоме. Его экспрессия в эпителии дыхательных путей исправляет дефектную регуляцию хлоридного канала в клетках эпителия. Прямой анализ CFTR-гена показал, что около 2/3 всех CF-хромосом несут специфическую мутацию ΔF508 на экзоне 10, а на остальной 1/3 имеется большое число последующих мутаций. В настоящее время их известно более 170 [40]. Прямой анализ данного гена имеет большое значение для пренатальной диагностики носительства. Наличие ΔF508, гомозиготы или комбинация с другими CFTR-мутациями доказывают диагноз «МВ». Затрудняет постановку диагноза лишь то, что еще не все CFTR-мутации охарактеризованы, а также различие клинического течения МВ и влияния на них экзогенных факторов. Так же как при ААТгене, имеется возможность прямого трансфера и экспрессии CFTR-гена в эпителий дыхательных путей. Сначала эту попытку предприняли на моделях животных, а затем у больных тяжелым МВ. По этому же принципу провели первые клинические испытания по генной терапии [11].
Ген NADPH-оксидазного комплекса
Более 30 лет назад, сразу после описания ААТ, было представлено генетическое заболевание, вызванное нарушением антимикробной защитной функции фагоцитов, которое было названо «ХГЗ». Больные страдают повторными бактериальными инфекциями, заканчивающимися в конце концов летально с формированием в легком множественных гранулем. Исследование состояния фагоцитов и метаболизма нейтрофилов у этих пациентов обнаружило, что эти клетки, в отличие от нормальных нейтрофилов, не продуцируют супероксидный радикал (О2-), и, таким образом, фагоцитированные бактерии не убиваются. Для строительства действенного антимикробного кислородного радикала необходим O2- оксидредуктазный энзим или NADPH-оксидаза, находящаяся в плазматической мембране фагоцитов. Первичным продуктом NADPH-оксидазы является супероксидный радикал.
Было показано, что это заболевание наследуется по аутосомно-рецессивному или Х-сцепленному рецессивному типу. Это означает, что заболевание генетически гетерогенно и обусловлено как минимум дефектами двух разных генов: один — Х-сцепленный, другой — аутосомный. Открытие того, что Х-сцепленная форма связана с отсутствием гемопротеина — цитохрома b558, позволяет предположить, что эта форма представлена Х-сцепленно кодированной компонентой — NADPH-оксидазным комплексом. В 1986 г.
ген для Х-хромосомной, цитохром-b558-негатив- ной формы заболевания ХГЗ (Xb-) был клонирован в соответствии с его хромосомной локализацией [47]. Ген кодирует тяжелую цепь цитохрома b558. Позже была разъяснена основа цитохром-b558-ак- тивной формы ХГЗ (AB) с дефектом в α-цепи гена. Помимо этих двух форм, при которых отсутствует мембранный цитохром в одной из цепей, открыли аутосомно-рецессивную цитохром-b558-пози- тивную форму синдрома периферического воспаления (AB+), связанную с отсутствием второго, цитозольного компонента NADPH-оксидазного комплекса [34]. ХГЗ — очень редкое заболевание, поэтому методы генной терапии для него не разработаны, хотя теоретически терапия его возможна методом трансфера гена.
Поиск новых генов риска бронхолегочных заболеваний
Наши знания о заболеваниях бронхолегочной системы, полученные различными методами исследования в сочетании с молекулярно-генети- ческими методами поиска генов риска, помогают лучше сориентироваться в патогенезе этих заболеваний. Выделяют следующие механизмы.
•Нарушение протеиназно-антипротеиназного равновесия в легком, в результате чего нарушается структура легочной ткани (через протеолитические энзимы). Прототипом этого довольно частого генетического дефекта про- теазно-антипротеазного равновесия служит описанный выше ААТ-дефицит.
•Генетическая регуляция продукции иммуноглобулина Е (IgE) и поиск отвечающих за нее генов. У больных атопией эта регуляция нарушена, что имеет большое значение в патогенезе БА и других подобных заболеваний.
•Нарушение антимикробных механизмов защиты в легком. Расшифровка генов, отвечающих за это нарушение, приведет к лучшему пони-
манию патогенеза болезней и новым терапевтическим возможностям.
Внутрилегочное протеиназно-антипротеиназное равновесие. В основе патогенеза ХОБЛ лежит про- теиназно-антипротеиназная теория, что было показано в экспериментах на животных и у людей. Известно, что протеиназы в легком обладают сильным поражающим механизмом, если неадекватно ингибируются их ингибиторами антипротеиназами. Так, ААТ обеспечивает около 90% антиэластазной активности легкого, а дефицит ААТ — это прототип генетического субтипа ХОБЛ, обусловленного протеиназно-антипротеиназным дисбалансом. В этом случае антагонисты нам известны: это нейтрофильная эластаза и ААТ. О физиологических функциях многих других внутрилегочных протеиназ и их ингибиторов известно очень мало. Однако в последние годы изучены некоторые протеиназы и антипротеиназы (табл. 1.6, 1.7).
42

Анатомия и онтогенез респираторной системы
Таблица 1.6. Протеиназы
Обозначение |
|
Хромосомный |
|
Молекулярная |
|
Происхождение |
Субстрат |
Ингибиторы |
|
|
|
регион |
|
масса, kDa |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Серинэластаза |
|
19p |
|
— |
|
АГН |
Эластин, |
ААТ |
|
|
|
|
|
|
|
|
фибронектин |
|
|
|
|
|
|
|
|
|
и др. |
|
|
|
|
|
|
|
|
|
|
|
|
Протеиназа-3 |
|
19р |
|
— |
|
АГН |
Эластин |
Неизвестный |
|
|
|
|
|
|
|
|
|
|
|
Азурофилин |
|
19р |
|
— |
|
АГН |
Эластин |
Неизвестный |
|
|
|
|
|
|
|
|
|
|
|
Катепсин G |
|
14q 11.2 |
26 |
|
АГН |
Эластин |
α1-Антихимотрип- |
||
|
|
|
|
|
|
|
|
|
син (ACT) |
Металлоэластаза |
|
— |
|
— |
|
Альвеолярные |
Эластин |
TIMP и |
|
|
|
|
|
|
|
макрофаги |
|
|
α2-макроглобулин |
|
|
|
|
|
|
(новый синтез) |
|
|
|
Металлоколлагеназа |
|
— |
|
— |
|
Специфические |
Коллаген |
TIMP и |
|
|
|
|
|
|
|
гранулы |
|
|
α1-макроглобулин |
|
|
|
|
|
|
нейтрофилов |
|
|
|
Примечание: АГН — азурофильные гранулы нейтрофилов; TIMP — тканевый ингибитор металлопротеиназ. |
|
||||||||
Таблица 1.7. Антипротеиназы |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
Обозначение |
|
Хромосомный |
|
Молекулярная |
|
Клетки |
|
|
Связывание |
|
|
регион |
|
масса, kDa |
|
происхождения |
|
|
|
|
|
|
|
|
|
|
|
|
|
ААТ |
|
14q 31–32 |
|
52 (12) |
|
Гепатоциты и |
|
Эластазы |
|
|
|
|
|
|
|
альвеолярные |
|
|
|
|
|
|
|
|
|
макрофаги |
|
|
|
|
|
|
|
|
|
|
|
|
|
ACT |
|
14q 31–32 |
|
68 (26) |
|
Гепатоциты и |
|
Катепсин G, химазы ТК |
|
|
|
|
|
|
|
альвеолярные |
|
|
|
|
|
|
|
|
|
макрофаги |
|
|
|
|
|
|
|
|
|
|
|
|
|
α2-Макроглобулин |
|
12p 12–13 |
|
720 |
|
Гепатоциты и |
|
Широкий спектр |
|
|
|
|
|
|
|
альвеолярные |
|
человеческих и |
|
|
|
|
|
|
|
макрофаги |
|
бактериальных протеиназ, |
|
|
|
|
|
|
|
|
|
вирусов, бактерий |
|
Антилейкопротеиназа |
|
— |
|
— |
|
Клетки бронхиального |
Эластаза, катепсин G |
||
|
|
|
|
|
|
эпителия |
|
|
|
|
|
|
|
|
|
|
|
|
|
Примечание: в скобках указан молекулярный вес углеводных цепей в общем молекулярном весе гликопротеинов.
Активированные нейтрофилы высвобождают кроме серинэластазы еще и катепсин G, про- теиназу-3 и азурофилин из азурофильных гранул (см. табл. 1.6), и все четыре энзима поражают эластическую структуру легкого [17]. Активированные нейтрофилы аккумулируются в легких курильщиков, так же как у пациентов с ХОБЛ. Кроме этих «эндогенных» протеиназ при инфекциях бронхиального дерева из разрушенных бактерий высвобождаются высокоактивные бактерийспецифические протеиназы, которые принимают участие в разрешении рецидивирующего воспаления при ХОБЛ. Такие защитные функции в легких осуществляются антипротеиназами (см. табл. 1.7).
Благодаря накопленным знаниям с помощью молекулярно-генетических методов были изучены еще два потенциальных гена риска для заболеваний легких — ACT и α2-макроглобулин-ген (А2М).
ACT — это ингибитор серинпротеиназы с до конца не известной физиологической функцией у человека. Это гликопротеин острой фазы,
который ингибирует химазу ТК и катепсин G (см. табл. 1.7). Он кодирован геном, принадлежащим к генной семье ААТ в регионе q31–q32 хромосомы 14 и удален от ААТ-гена не далее чем на 130 базовых пар [49]. По многим причинам его причисляют к потенциально новым генам риска респираторных заболеваний. Он ингибирует продукцию супероксида в нейтрофилах, регулирует их хемотаксис, индуцированный катепсином G, и стимулирует продукцию протеинов острой фазы после связывания с катепсином G [24]. Описан частичный ACT-дефицит среди шведского населения (практически у каждого 200го жителя). Гомозиготы этой дефектной аллели пока не найдены, а гетерозигота для дефектной аллели ассоциируется с нарушениями функции внешнего дыхания (ФВД) [39]. У некоторых пациентов с ACT-дефицитом были обнаружены сочетания криптогенного цирроза печени с ХОБЛ. Далее с помощью PCR-амплификации и прямого секвенирования были обнаружены мутации ACTгена, которые ассоциировались с аномалиями на других протеинах при ХОБЛ. Была установлена
43

Раздел 1
точечная мутация Pro229–Ala, затем Len55Pro молекулярного базиса первых известных дефектов фенотипа ААТ. Эта мутация была обнаружена в семье с тяжелым течением ХОБЛ в трех поколениях.
А2М — это ингибитор протеиназ, который появляется в сыворотке человека в высоких концентрациях (8–10% общего количества протеинов). У человека он не является протеином острой фазы, синтезируется в гепатоцитах, альвеолярных макрофагах и фибробластах. А2М ингибирует намного более широкий спектр протеолитических энзимов, чем ААТ и ACT, а именно все протеиназы человеческого и бактериального происхождения [4]. Человеческий А2М-мономер кодируется геном на хромосоме 12, который принадлежит генному комплексу с еще как минимум двумя близкородственными генами [12]. А2М-ген крысы и человека клонирован и частично секвенирован, так же как и А2М-рецептор. В то время как уже известны его структура и механизмы влияния in vitro, физиологические функции его in vivo пока не до конца ясны. Он ингибирует практически все протеиназы и рост Pseudomonas aeruginosa, Serratia marcenscens, Influenzaviren, что соответствует функциям примитивной защитной молекулы и универсального ингибитора протеиназ. Факт его локальной и регулируемой экспрессии в легких благодаря альвеолярным макрофагам позволяет предположить, что при определенных условиях
внем испытывают нужду высвобождаемые через рецептор протеиназы для процесса комплексирования и элиминации. А это, в свою очередь, должно приводить к тому, что хронические воспалительные процессы и структурные изменения в легких, согласно протеиназно-антипротеиназной теории, должны уменьшаться. В настоящее время отсутствуют исследования А2М, подтверждающие конкретные клинические проявления генетического дефекта этого протеина. Имеются данные о больных ХОБЛ и с частичным А2М-дефицитом
вкомбинации с дефектом субклассов иммуноглобулина G (IgG), хотя неясно, причиной или следствием тяжелого состояния больных является А2М-дефицит.
Генетическая регуляция продукции иммуноглобулина Е
Атопия — это сложное болезненное состояние, при котором клинической манифестацией являются БА, нейродермит, аллергический ринит (АР) с наличием аллергенспецифических IgEАТ и повышенного содержания IgE в сыворотке. Хотя генетические элементы атопии до сих пор полностью не изучены, семейная агрегация атопий четко прослежена [51]. Дети с родителями без атопии отягощены в 15% наблюдений, с одним родителем с атопией — в 30%, а с двумя — в 50% случаев. Появление специфических
IgE ассоциируется с главным комплексом гистосовместимости (MHC). Среди людей с уровнем IgE ниже 60 МЕ/ml (примерно 70% населения) атопия встречается приблизительно в 5%, при уровне 200–450 МЕ/ml (10% населения) — в 40%, а выше 450 МЕ/ml — практически в 100% случаев. Общий уровень сывороточного IgE контролируется одним или многими генами, что было доказано в экспериментах на животных. Во многих семьях с атопией проводились анализы сцепления и была доказана тесная связь IgE-ответа с маркером D11S19 на хромосоме 11q [8]. Для выявления более точной локализации пытаются применять позиционное клонирование.
Защитные антибактериальные механизмы легких
Система защиты легких сложна и до сих пор не до конца понятна. Однако два аспекта: антимикробные протеины фагоцитов и селективные состояния недостатка иммуноглобулинов — являются очень интересными.
Фагоциты (нейтрофилы и эозинофилы, моноциты и макрофаги) — эффекторные клетки антимикробной защиты. Они уничтожают микроорганизмы посредством оксидативных и неоксидативных механизмов.
ХГЗ, описанное выше, — хорошо изученный генетический дефект с такими механизмами. Оно обусловлено дефектом энзима NADPH-оксидазы, что делает невозможным адекватную оксидативную защиту от микроорганизмов. К неоксидативным механизмам относится высвобождение антимикробных протеинов фагосом, однако генетические дефекты таких протеинов до сих пор не выяснены.
Синтез и функции иммуноглобулинов классов IgG и IgA, так же как и IgE, — процесс комплексный, а интерпретация клинических проявлений иммунодефицита, который мы наблюдаем у большинства пациентов с ХОБЛ, очень сложна. Так, полный недостаток иммуноглобулинов A (IgA) — частый иммунный дефект, который ассоциируется с синопульмональными инфекциями вирусного и бактериального генеза и часто наблюдается у больных атопией. Генетическая основа такого дефекта абсолютно неясна, как и IgG (субкласс IgG2, IgG4). Такой же минимум генетической информации имеется о других классах иммуноглобулинов.
Первичная цилиарная дискинезия
Первичная цилиарная дискинезия (ПЦД) представляет собой наследственную гетерогенную патологию с аутосомно-рецессивным типом наследования. Вследствие несостоятельности мерцательного РЭ и нарушения его цилиарной функции у больных может развиваться целый
44

Анатомия и онтогенез респираторной системы
спектр поражений различных внутренних органов, однако более всего страдает респираторная система, о чем свидетельствует наличие рецидивирующих бронхитов и затяжной пневмонии. Считается, что 50–60% ПЦД приходится на синдром Картагенера [7]. Впервые этот синдром описан в 1904 г. А.К. Зивертом, но более детальное описание данной патологии, ее семейных форм было представлено М. Картагенером в 1933 г. [28]. Синдром Картагенера (синдром Зиверта– Картагенера) — это триада, включающая в себя БЭ, синуситы и situs viscerus inverus. В 1970-х годах R. Eliasson (1977) и B. Afzelius (1978) выявили у больных с данной триадой дефект строения аксонем ресничек мерцательного эпителия слизистой оболочки бронхов [41]. Таким образом, в основе ПЦД лежит генетически детерминированное изменение его ультраструктуры. В настоящее время при ПЦД их описано свыше 20 [5]. Применяя метод кандидатного картирования, хромосомы 3p, 4q, 5p, 7p, 8q, 10p, 11q, 13q, 15q, 16p, 17q и 19q обозначили как геномные регионы, которые могут быть ассоциированы с ПЦД [18]. Анализ семей
снаследственной декстрапозицией указывает на связь с хромосомами 8q и 19q. При исследовании по сцеплению фенотип синдрома Картагенера в арабских семьях был картирован в области 19q (19q13.3-qter) [16]. Облигатными признаками патологии считаются отсутствие или недоразвитие динеиновых ручек, отсутствие радиальных спиц, нексиновых связок, нарушение числа дублетов и синглетов. Динеиновые ручки являются носителями АТФ-активности, превращающей химическую энергию АТФ в механическую энергию движения ресничек. В последние годы динеиновые дефекты связывают с мутацией в области короткого плеча хромосомы 9 (9р21–р13), а причину «дефицита» динеиновых ручек — с 8q и 16pter [21]. Утрата динеиновых ручек, что происходит в 70–80% наблюдений ПЦД, приводит к неподвижности ресничек. В 2002 г. H. Olbrich и соавт., обследовав 7 человек из 6 семей с синдромом Картагенера, обнаружили мутацию в гене DNAH5 (axonemal heavy chain dynein type 5), расположенном в геномном регионе 5p15–p14. Другая группа исследователей показала мутацию в гене DNAH11 (axonemal heavy chain dynein type 11). Также при изучении гена DNAH11, располагающегося в области 7р21 хромосомы, были идентифицированы мутация в 82-м экзоне и гомозиготная нонсенс-мутация (R2852X) [3]. У пациента с синдромом Картагенера была установлена мутация в гене DNAI1 (IC78) (9p21–p13) [30]. В одном из последних исследований 30 семей
сПЦД было установлено 33 новых (12 нонсенс-, 5 сплайсинг-, 8 миссинг-мутаций и 8 мутаций со сдвигом рамки считывания) и 2 известные ранее мутации в гене DNAH5 [36]. Интересно, что у 6 (32%) из 19 обследованных семей с ПЦД из Северной Америки была обнаружена новая основная мутация в 10815delIT.
Анализ последних исследований по поиску генетических маркеров генов бронхолегочной патологии
ВЮжной Атлантике есть один маленький остров — Тристан да Кунха, который считается «островом астматиков». Каждый третий житель острова страдает БА, что абсолютно не связано с факторами внешней среды: просто из 15 первых поселенцев — жителей острова — трое страдали БА. Интересно, что эти наблюдения не связаны
ссостоянием атопии, так как частота позитивных аллерготестов среди жителей острова не выше, чем в других частях планеты [43]. Скорее всего, жители острова унаследовали состояние гиперреактивности бронхов.
Английское проспективное исследование показало, что среди рожденных в 1946 г. детей по достижении ими 4-летнего возраста у 6,2 на 1000 человек была диагностирована БА [54]. Дальнейшее наблюдение за этими людьми показало, что среди их детей уже было 18,9 на 1000 больных БА детей. Ссылка на лучшую диагностику БА в последние годы вряд ли может объяснить такой резкий подъем.
Исследование стационарных пациентов в Германии показало, что позитивный по БА семейный анамнез встречается при экзогенно-ал- лергической форме БА в 81% наблюдений, когда больные указывают на повторные случаи БА в семье (у родителей, братьев, сестер или детей) [43]. А среди пациентов с терапевтической патологией в Германии в 20% наблюдений имеется указание на семейную отягощенность по БА.
Считается, что наследуется состояние гиперреактивности бронхиальной системы, но то, как реализуется эта гиперреактивность, во многом зависит от факторов окружающей среды. Факторами риска развития БА у детей раннего возраста считаются:
•семейный анамнез (БА у матери — действенно больший фактор риска, чем у отца, но наибольший риск — при наличии БА у обоих родителей);
•указание на атопический статус, например атопическая экзема (нейродермит);
•повышенный уровень IgЕ в крови пуповины;
•наличие эозинофилов в крови 9-месячного ребенка;
•позитивные кожные аллерготесты у ребенка после 6 лет.
Вто же время имеются совершенно противоположные данные: так, в Швеции при исследовании 7000 монозиготных (однояйцовых) близнецов показано, что, несмотря на абсолютную их идентичность, только 20% пар страдают астмой [13]. Возможно, этот «астматический вклад» наследуется по какому-то единичному гену аутосо- мально-доминантным путем и клиническое про-
45

Раздел 1
явление во многом детерминировано факторами окружающей среды.
Некоторые сильные генетические компоненты БА уже известны. По данным эпидемиологических исследований, патогенез этого заболевания в значительной степени детерминирован генетическими факторами [45]. При этом результирующая клиническая картина заболевания у пациентов проявляется под воздействием экзогенных факторов — это так называемая фенотипическая манифестация генетической предрасположенности.
Клиницисты всегда обращали внимание на семейную агрегацию этого заболевания; не хватало только соответствующих молекулярно-гене- тических технологий. Современные технологии позволили изучить роль генов в патогенезе БА, которые в настоящее время изучаются структурно и функционально, вплоть до их молекулярных деталей. Поиск естественно встречаемых дефектов в кандидатных генах наряду с анализом трансгенных экспериментальных моделей и рекомбинантных штаммов животных — это многообещающая возможность приблизить решение проблемы комплексной генетической предрасположенности к заболеваниям.
Предшествующие исследования на семьях больных не подтвердили гипотезу о простом менделевском наследовании БА в целом, что определило продолжение активных генетических исследований [1].
ВАвстралии для определения кандидатного гена БА провели мутационный скрининг гена IFN-γ [19]. Ген IFN-γ расположен на хромосоме 12 — в регионе генома, связанном с БА. Было проведено исследование ПЦР геномной ДНК соответствующих регионов у 265 пациентов из двух популяций — Западной Австралии и Венесуэлы. Мутации в гене IFN-γ оказались недостоверными, что не позволило считать этот ген маркером БА в этих популяциях.
ВАвстралии изучали семейную агрегацию БА среди местного населения [26]. Пробандами выступали люди, рожденные в 1961 г. Всего изучено 7394 семьи с 41 506 членами семей. Главной задачей этого популяционного исследования было выяснение вопроса о наличии действительной семейной агрегации БА в семьях, ее связи с факторами внешней среды и генетическими факторами. Были применены и проанализированы различные модели наследования БА — регрессивное моделирование в сочетании с эффектом «родитель–ребенок», олигогенное моделирование, неменделевское распределение наследования, менделевская модель, кодоминантная модель, доминантная модель, а также рассчитаны соответствующие коэффициенты наследования. Показано, что, во-первых, скорее всего, имеется не один главный локус, ответственный за БА, во-вторых, гены БА в популяции наследуются
кодоминантно, а факторы окружающей среды в развитии БА у жителей Австралии не являются доминирующими; тем самым был доказан вклад генетических факторов в развитие БА.
ВГермании изучили ассоциацию высоких уровней сывороточного IgE с HLA-DR и маркерами на хромосоме 5q31 и хромосоме 11q13 [53]. Предварительные исследования выявили связь между общими сывороточными концентрациями IgE и областью хромосомы 5q31, а также с локусом атопии хромосомы 11q13. Этим данным противоречили другие, указывающие на то, что управляемая антигенами продукция IgE контролируется
восновном комплексом генов II класса MHC. Настоящее же исследование проанализировало ассоциацию между фенотипом высоких уровней сывороточного IgE и шестью микросателлитными маркерами хромосом 5q31 и 11q13, а также и HLA-DRB1 в рандомизированной выборке взрослых. 129 человек с уровнем IgE выше 200 МЕ/мл и 266 контрольных участников с IgE ≤200 МЕ/ мл были генотипированы на 5 микросателлитных маркеров хромосомы 5q31, а именно на D5S436, D5S393, D5S210, IL-4 и -9; на один микросателлит хромосомы 11q13, а именно FCERIB, и все они типированы на HLA-DRB1. Результаты анализировали в соответствии с тестом Фишера. Показано, что ни один из маркеров не ассоциировался достоверно с высоким уровнем IgE, хотя была найдена слабая ассоциация высокого уровня IgE с генами IL-9, FCER1B и HLA-DRB1 при сравнении с контролем.
Генетический контроль взаимосвязи БА с уровнем сывороточного IgE изучали в изолированной популяции Финляндии [31]. Учитывая, что геномный регион хромосомы 5 предварительно применялся для изучения уровня IgE, а также бронхиальной гиперреактивности (БГР), т.е. наследственной предрасположенности к астме, решили изучить данный регион. Чтобы подтвердить эту связь, ограничить исследуемую область генома, и использовали изолированную популяцию. 16 полиморфных маркеров, включая гены IL-4 и -9, были упорядочены и генотипированы в 157 ядерных семьях. В результате не было обнаружено генетической связи в сиблинговых и кузиновых парах между этими маркерами на хромосоме 5q как с сывороточным уровнем IgE, так и с БА. Также провели анализ ассоциаций гаплотипов. При этом установили, что в данной изолированной финской популяции вариации аллелей хромосомы 5q31 не участвуют в наследовании уровня сывороточного IgE и развития БА.
Вштате Колорадо (США) с 1998 г. проводится большое национальное исследование «Кандидатные гены и БА» [46]. Изучают вопросы патофизиологии БА, генетики и физиологии цитокинов, физиологии генов, генетики IL-4, физиологии IL-4, генетики маркеров связи, генетического полиморфизма. Результаты этого ис-
46

Анатомия и онтогенез респираторной системы
следования должны внести ясность в решение вопроса о генах БА.
Вштате Аризона (США) проведено исследование связи циркулирующих в крови эозинофилов с маркерами хромосомы 5q [38]. Хорошо известно, что эозинофилия сопряжена с риском развития БА, при этом ее генетическая регуляция не изучена. Изучались связи между циркулирующими эозинофилами и 9 маркерами хромосомы 5q31–33
усиблинговых пар (sib-pair analysis). Показано, что контролирующие эту связь локусы могут быть представлены на этой хромосоме, контролирующей циркуляцию эозинофилов пропорционально общему числу клеток белой крови.
В1999 г. в Германии создан банк данных, содержащий 88 исследований по связям и 72 исследования мутаций при БА и аллергии, который постоянно пополняется [24].
Вто время как многие генетические исследования по БА сфокусированы на молекулах и генах, которые участвуют в процессах воспаления и иммуномодуляции, позиционное клонирование гена ADAM33 открыло новые возможности в респираторных исследованиях. Ученые, открывшие роль гена ADAM33 при БА, описывают, как «длинная и путаная дорога сначала завела» их «так далеко — к позиционному клонированию гена ADAM33, а потом вернулась обратно к истокам — его возможной роли в формировании бронхиального древа» [27].
Табачный дым и пылевые загрязнения (в том числе и профессиональные) — известные факторы риска ХОБЛ. Хотя курение в данном случае — важнейший фактор риска, только у 15% курильщиков развивается эта болезнь. Семейные исследования, проведенные в ФРГ, показали, что для развития ХОБЛ необходима важная полигенная компонента. Исследования 252 пар среди монозиготных близнецов и родных братьев-сестер курильщиков подтвердили повышенную частоту риска развития ХОБЛ по сравнению с пробандами, не имеющими родных сибсов. В настоящее время можно с уверенностью говорить о роли гена, подавляющего
α1-протеазу в развитии ХОБЛ (API-ген). Однако API-ген содержит огромное число секвенц-ва- риаций, из которых за данный функциональный дефект отвечает точечная мутация на 5-м экзоне. По соседству с API-геном располагается ген, подавляющий протеазу ACT, две секвенц-вариации которого также «вносят вклад» в развитие ХОБЛ. Другие кандидатные гены ХОБЛ — гены коллагеназы (MMP-1) и ген желатиназы В (MMP-9). Генами-кандидатами воспалительного процесса при ХОБЛ являются гены системы лимфогематопоэза — ген фактора некроза опухоли α (TNF-α) и гены интерлейкинов (IL). Кандидатными генами для компенсации оксидативного стресса являются гены супероксиддисмутазы и гены микросомальной эпоксидгидролазы [55].
Ген ткани ингибитора металлопротеиназ (TIMP-2) существенно предрасполагает к разви-
тию ХОБЛ, причем исследования в этом направлении настолько интенсивны, что практически ежегодно идут новые сообщения об изученных полиморфизмах [20].
Интересны данные по изучению генетики легочной гипертензии. Семейную идиопатическую легочную гипертензию изучали как в спорадических случаях, так и в семьях с накопленными случаями данного заболевания. Показано, что болезнь наследуется аутосомно-доминантным путем. Дефект находится в области гена костного морфогенетического рецептора протеина типа II, который кодируется как трансформированный ростовой фактор β. Показано также, что чисто спорадических случаев данного заболевания не существует, так как при тщательном генетическом семейном анализе обнаружено, что в основе заболевания всегда существует мутация данного гена. Анализ наблюдений ассоциации идиопатической легочной гипертензии с наследственной геморрагической телеангиэктазией показал, что в развитие идиопатической легочной гипертензии может быть также вовлечена мутация гена акти- вин-рецептор-киназы 1. В связи с полученными данными развиваются подходы для генетического консультирования и помощи в семьях с повторными случаями данного заболевания.
Эволюция — это логичное объяснение межпопуляционных различий специфических аллельных частот, если вовлеченные гены имеют связанные друг с другом функции, гетерогенные аллели имеют схожие функциональные последствия, вовлеченные гены не связаны хромосомально между собой [33]. Такая комбинация факторов может привести к биологически логичным, повышающим стойкость к выживанию взаимовлияниям генов и окружающей среды. Было показано существование постоянства между частотами аллелей в генах, которые связаны с иммунным ответом Т-хелперов 2 (Th-2) у людей с различным происхождением, но проживающих в климатически сходных регионах. Ответы Th-2 развились у млекопитающих для противостояния паразитарной инфекции, особенно против гельминтов. Современный человек произошел из тропической Африки, где он постоянно сталкивался с гельминтами. Сравнительно недавно человечество мигрировало в более холодные и сухие климатические зоны, которые для большинства гельминтов оказались неблагоприятными, поэтому у них появились сложности с размножением. Генетическая тенденция к сильным ответам Th-2 привела к лабильности здоровья. С одной стороны, человек приобрел устойчивость
кпаразитарной инфекции, а с другой, у него повысилась врожденная предрасположенность
каллергическим и атопическим заболеваниям. В данных процессах участвуют специфические аллели IL-4 и его рецепторов, IL-10 и -13, β-цепь рецептора IgЕ, адренергический рецептор β1, α-цепь TNF. Данные полиморфизмы, специфичные для
47

Раздел 1
популяции, имеют большое значение для развития заболеваний. Высокая частота БА у мигрантов из тропических регионов, переехавших в более умеренный климат, связана именно с этими процессами. Следует отметить высокую вероятность того, что повышающаяся ассимиляция в западное общество 2 млрд человек из тропиков может привести к быстрому росту заболеваемости БА, поскольку такое население уже имеет высокую генетическую предрасположенность к аллергическим заболеваниям.
Большое распространение бронхолегочной патологии в развивающихся странах — предмет для беспокойства в будущем [37]. Поэтому в настоящее время на первый план для исследователей выдвигается изучение генов, взаимодействующих с окружающей средой. Окружающая среда — это ключ к пониманию генетики. Генетические ре-
зультаты не могут иметь большого смысла, если окружающую среду не брать во внимание. Ученые постепенно приходят к выводу, что изучение генотипов только в изолятах может завести в тупик. С другой стороны, целый ряд генетических факторов, сцепленных с определенным заболеванием индивидуума, приводит к развитию болезни в том случае, если конкретные пусковые механизмы окружающей среды тоже существуют. Это и объясняет тот факт, почему так трудно определить генетические маркеры для мультифакториальных заболеваний, и подчеркивает необходимость мультидисциплинарных подходов для объективной оценки генетических данных.
Список литературы
См. 
48
Раздел
2физиологияРеспираторная
2.1. Респираторная функция легких: вентиляция, газообмен и кровообращение
З.Р. Айсанов, Е.Н. Калманова, А.В. Черняк, С.Ю. Чикина, Ж.К. Науменко
В настоящем разделе рассматриваются вентиляция и кровоток, лежащие в основе газообмена. Хотя легкие имеют целый ряд нереспираторных функций, таких как метаболическая и удаление нежелательных компонентов из циркуляторного русла, тем не менее дыхательная функция является основной. При болезнях органов дыхания часто нарушаются вентиляция, кровоток и газообмен, что может приводить к дыхательной недостаточности (ДН) и смерти.
Вентиляция
Дыхательные пути представляют собой последовательность разветвляющихся трубок. По мере деления они становятся уже и короче, количество их возрастает по мере проникновения в легкие. Все эти бронхи, включая терминальные бронхиолы, представляют собой проводящие дыхательные пути. Их функцией является функция проведения в газообменные отделы легких. Поскольку проводящие дыхательные пути не содержат альвеолы, то они представляют собой анатомическое мертвое пространство.
Каждой респираторной бронхиоле сопутствует респираторная единица — ацинус. Каждая терминальная бронхиола делится на респираторные бронхиолы, от стенок которых отпочковываются единичные альвеолы. Далее идут альвеолярные ходы — структуры, полностью связанные с альвеолами. Этот отдел легких, где присутствуют альвеолы, называется респираторной зоной. Отдел, расположенный дистально по отношению к терминальным бронхиолам, называется еще переходной или респираторной зоной, так как отделы респираторных бронхиол, где альвеолы отсутствуют, не выполняют респираторной функции. Дистанция от терминальной бронхиолы до наиболее дистальных альвеол составляет всего лишь 5 мм, тем не менее респираторная зона составляет большую часть легких (ее объем около 2–3 л).
Сегодняшние представления о морфологии дыхательных путей с функциональной точки зрения во многом базируются на работах Weibel. В этих работах измерялись количество, длина, ширина и углы деления дыхательных путей. Были предложены модели, которые хотя и являются идеализированными, тем не менее они делают возможным различные виды анализов респираторных кривых (таких как кривая «давление–объем»).
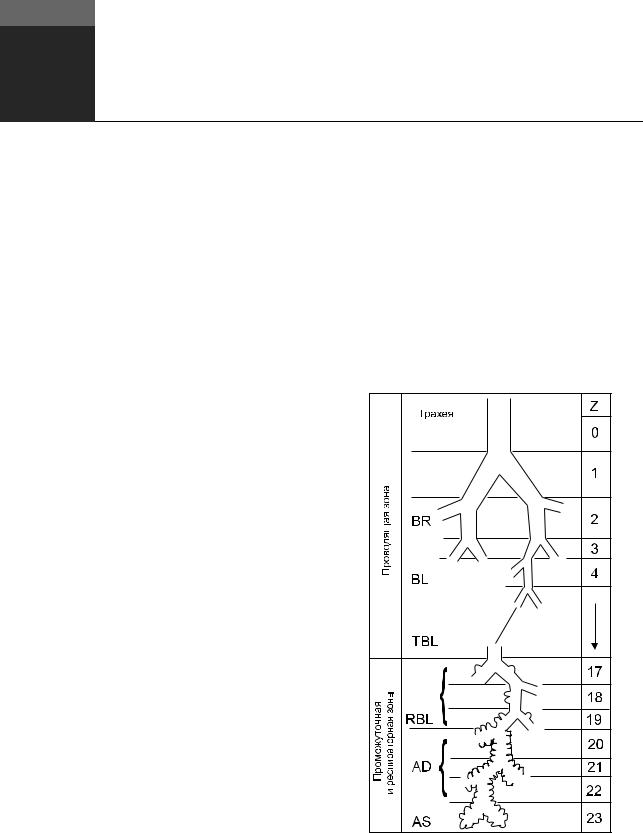
Наиболее часто используется так называемая идеализированная модель А (рис. 2.1), в которой первые 16 генераций, включая терминальные дыхательные пути, составляют проводящую зону.
Рис. 2.1. Дыхательные пути человека в соответствии с мо- |
делью A. Weibel: AD, AS — альвеолярные мешочки; BL — |
бронхиолы; BR — бронхи; RBL — респираторные бронхи- |
олы; TBL — терминальные бронхиолы; Z — генерации ды- |
хательных путей. RBL, AD и AS формируют промежуточную |
и респираторную зоны (Weibel E.R. Morphometry of the Hu- |
man Lung. Berlin: Springer-Verlag, 1963) |
49