

Болезнь Гентингтона (хорея Гентингтона, Хантингтона): общие сведения (0001)
Обратные ссылки
Хорея Гентингтона ( MIM 143100 ) - одно из самых тяжелых прогрессирующихнейродегенеративных наследственных заболеваний головного мозга . Хорея (chorea; от греческого слова "choreia" - пляска) - форма гиперкинеза, характеризуется непроизвольными, быстрыми, нерегулируемыми движениями, возникающими в различных мышечных группах.
Его распространенность составляет около 10:100000. Отличительные признаки - хорея ирасстройства поведения . Заболевание может начинаться с любого из этих симптомов или с обоих сразу. Болезнь Гентингтона может развиться в любом возрасте - как в детстве, так и в 70 лет и старше, но чаще первые симптомы появляются в 30-50 лет.
Хорея начинается исподволь. Первые признаки хореи Гентингтона проявляются в возрасте 25-50 лет, реже в детском возрасте. Мужчины болеют чаще, чем женщины; первыми симптомами могут быть неусидчивость , суетливость движений , что не расценивается больным и его родственниками как заболевание. Со временем, однако, двигательные нарушения нарастают и могут привести к инвалидности. Характерны частые, внезапные, неритмичные судорожные движения конечностей или туловища. Возможны спазмы лицевой мускулатуры , всхлипывания ,нарушения артикуляции . Страдает координация движений при ходьбе : походка становится "танцующей" (хореической) . Память остается сохранной вплоть до поздних стадий заболевания, однако внимание , мышление и исполнительные функции нарушаются уже в самом начале заболевания. Часто наблюдаются депрессия , апатия , отчужденность , раздражительность ,периодическая расторможенность . В некоторых случаях развиваются бред и навязчивые состояния , в связи с чем сначала ошибочно диагностируется шизофрения .
Продолжительность заболевания различна, но в среднем составляет 15 лет. При раннем начале (до 20 лет) заболевание сопровождается ригидностью , атаксией , когнитивными нарушениями и прогрессирует быстрее (средняя продолжительность составляет 8 лет). Эпилептические припадки редко возникают при болезни Гентингтона с поздним началом и часто - при ранней форме.
Обычно хорея Гентингтона проявляется на 4-5-ом десятилетии жизни прогрессирующимхореоатетозом , который сопровождается выраженными психическими расстройствами (деменцией , депрессией с нередкими суицидальными попытками, нарушениями эмоционального контроля с частыми вспышками раздражения и агрессии ). Иногда проявляется в юношеском возрасте нарастающим акинетико-ригидным синдромом .
Заболевание характеризуется преимущественно поражением стриатума . На развернутой стадииКТ или МРТ выявляют атрофию хвостатых ядер , образующих латеральные стенки боковых желудочков ( рис. 367.4 ). При микроскопическом исследовании выраженных изменений (таких, как амилоидные бляшки и нейрофибриллярные включения при болезни Альцгеймера ) не находят. Характерны глиоз и гибель преимущественно ГАМКергических нейронов в хвостатом ядре и скорлупе . Холинергические нейроны остаются относительно сохранными. В базальных ядрах значительно снижаются концентрации ГАМК и глутаматдекарбоксилазы - фермента, участвующего в синтезе этого медиатора. Уменьшены концентрации и других медиаторов, в том числе вещества Р и энкефалинов . Магнитно-резонансная спектроскопия выявляет увеличение концентрации лактата в базальных ядрах . У животных введение 3-нитропропионовой кислоты, ингибитора сукцинатдегидрогеназы, вызывает состояние, напоминающее болезнь Гентингтона .
Ген болезни Гентингтона HD располагается в сегменте 4р16.3 , содержит повторяющиеся тринуклеотидные повторы ЦАГ и кодирует белок, называемый гентингтином . Этот белок содержится в нейронах различных отделов головного мозга; функция его неизвестна. Инактивация гомологичного гена у мышей при гомозиготном состоянии вызывает гибель зародыша; гетерозиготные особи фенотипически не отличаются от нормы. У мышей, у которых в этом гене увеличено число тринуклеотидных повторов, развиваются прогрессирующие двигательные нарушения. Увеличение числа повторов ЦАГ приводит к тому, что в кодируемом этим геном белке появляется длинный полиглутаминовый участок, что и может быть причиной заболевания. Полагают, что длинный полиглутаминовый участок нарушает связывание белков, а также другие процессы в клетках, например активность митохондрий . Сообщалось о нарушении связывания гентингтина с глицеральдегидфосфатдегидрогеназой . Кроме того, имеются данные об усилении апоптоза нейронов при этой болезни.
Болезнь Хантингтона представляет собой доминантную нейродегенеративную болезнь , вызываемую удлинением CAG триплетного повтора в участке гена, кодирующем N- концевую часть белка хантингтина , функция которого неизвестна. В норме длина этого повтора варьирует у разных индивидуумов от 11 до 34 триплетов. У больных его длина колеблется от 37 до более чем 100 кодонов. Этот дефект, по-видимому, оказывает свое влияние через измененный белок. Однако неясно, каким образом.
Одна из возможностей заключается в том, что удлинение полиглютаминового кластера в белке уменьшает его нормальную активность (механизм "потери функции"). Однако индивидуумы, у которых одна копия гена инактивирована не за счет удлинения повтора, а за счет повреждения структуры хромосомы в этом месте, хотя и имеют пониженное содержание "хантингтина", но не проявляют признаков болезни Хантингтона. Это противоречит механизму "потеря функции", хотя остается возможность так называемой негативной доминантности , при которой молекулы белка с утраченной функцией ингибируют функционирование нормального белка. (Обратите внимание, как разнообразны возможности повредить нам с вами: негативная доминантность достаточно широко распространена).
Другая возможность заключается в том, что белок с удлиненным полиглютаминовым участком сам приобретает новую вредную функцию.
Чтобы сделать выбор между двумя возможностями возникновения болезни вследствие мутации: "приобретение функции" или "потеря функции", мышиный гомолог гена был инактивирован путемген-таргетинга . см. Болезнь Хантингтона: исследование на трансгенных мышах
Выявление увеличения числа тринуклеотидных повторов ЦАГ в гене HD лежит в основе генодиагностики болезни Гентингтона. Если число повторов ЦАГ составляет 38 или более, то заболевание с возрастом разовьется неминуемо. Вероятность наследования аномального гена и, следовательно, развития болезни Гентингтона у детей таких больных составляет 50%. Чем больше тринуклеотидных повторов содержит ген, тем раньше начинается заболевание. Однако в большинстве случаев число повторов составляет 40-49, а в этих пределах зависимости между числом повторов и возрастом начала заболевания нет. По неясным причинам риск значительного увеличения числа тринуклеотидных повторов и развития ювенильной формы болезни Гентингтона (форма антиципации ) выше, если болен отец больного.
Прежде чем проводить анализ ДНК у здоровых лиц с высоким риском болезни Гентингтона, необходимо тщательное генетическое консультирование, поскольку положительные результаты анализа ДНК могут привести к тяжелой моральной травме. Подробные инструкции по генетическому консультированию и анализу ДНК имеются в литературе.
Медико-генетическое консультирование и анализ ДНК используются не только при определении риска болезни Гентингтона, но и в дифференциальной диагностике этого заболевания. Так, мутантный ген HD иногда находят при спорадической "старческой" хорее . При заболеваниях, напоминающих болезнь Гентингтона, таких, как шизофрения , доброкачественная семейная хорея , наследственные атаксии , абеталипопротеидемия и семейная болезнь Альцгеймера , число тринуклеотидных повторов не увеличено.
Специфического лечения нет. Двигательные нарушения и поведенческие расстройствауменьшаются под действием фенотиазинов , галоперидола или бензодиазепинов .
Болезнь Хантингтона неизлечима, но существует лечение, способное облегчить некоторые симптомы.[28]
Тетрабеназин был разработан специально для уменьшения тяжести симптомов болезни Хантингтона[29], был утвержден в 2008 году в США[30]. Нейролептики и бензодиазепиныпомогают уменьшить проявления хореи[22]. Амантадин и ремацемид находятся в стадии исследования, но показали положительные результаты[31]. Для облегчения гипокинезиии ригидности мышц назначают противопаркинсонические лекарства, для облегчения миоклонической гиперкинезии - вальпроевую кислоту[22].
Для устранения депрессии применяют селективные ингибиторы обратного захвата серотонина и миртазапин, а при психозах и нарушениях поведения назначаютатипичные антипсихотики[32].
В настоящий момент ведутся активные исследования по разработке способа лечения: Обнаружено потенциальное направление для лечения хореи Гентингтона
Арахнодактилия
Арахнодактилия описана в литературе впервые в 1896 г. известным французским педиатром Марфаном (Marfan) под названием долихостеномелия и до сих пор еще сплошь и рядом обозначается как болезнь Марфана. Настоящее же общепринятое наименование „арахнодактилия” предложил в 1902 г. Ашар (Achard). Это врожденное системное заболевание относится к числу сравнительно редких. Как и другие представители этой группы болезней, арахнодактилия встречается то неожиданно спорадически, то в качестве семейного заболевания, которому подвержено несколько членов одной и той же семьи из одного или разных поколений. Преимущественного поражения мужского или женского пола и здесь никто не отметил.
Арахнодактилия выражается в непомерном удлинении всех трубчатых костей, в особенности же наиболее периферически расположенных — фаланг рук и ног, пястных и плюсневых костей, но также и в изменениях черепа, скелета туловища. Ручки и ножки становятся поразительно длинными и узкими, вытянутыми вдоль длинных осей, и пальчики согнуты в тонких суставчиках. Это действительно порождает подобие с члениками паука, а отсюда и удачное, пусть и чисто внешнеописательное, название болезни — арахнодактилия (от древнегреческого арахнос — паук). Кроме изменений опорно-двигательного аппарата имеются еще обязательно очень характерные глубокие поражения других систем, в первую очередь изменения глаз и сердечно-сосудистые, что опять-таки говорит о том, что все врожденные системные так называемые костные болезни никоим образом не являются одним только костным патологическим процессом, а лишь костным или преимущественно костным выражением общего заболевания человеческого организма.
Патологический процесс при арахнодактшши начинается, несомненно, в ранней стадии внутриутробного развития и, стало быть, всегда уже выражен у новорожденного ребенка и в первый период жизни. Но совсем нередко болезнь проявляется и усиливается лишь при дальнейшем росте и развитии ребенка. Арахнодактилия — очень тяжелая болезнь с весьма неблагоприятным предсказанием, т. е. с очень высокой общей летальностью, и большинство детей гибнет в первые же годы жизни от любой привходящей, для них всегда опасной болезни. Лишь очень редко больные арахнодактилией остаются в живых до старшего или юношеского возраста, а тем более до зрелости и даже старости, и то это относится лишь к относительно весьма редким ее доброкачественным формам.
К обязательным симптомам арахнодактилии следует причислить прежде всего резко выраженную общую мышечную атрофию с атонией всей скелетной мускулатуры, типа врожденной амиотонии [myatonia congenita Оппенгейма (Oppenheim)]. Мы никогда ни при одном другом заболевании не видели ничего, отдаленно приближающегося к состоянию мышц у больных арахнодактилией. Мышцы конечностей на ощупь, можно сказать, просто как бы отсутствуют, они вообще не выделяются из-под кожи, они имеют в тяжелых случаях этого заболевания консистенцию буквально слизи. Это впечатление усугубляется еще тем обстоятельством, что кожа крайне истончена, а жировой клетчатки у страдающих арахнодактилией характерным образом вообще не бывает, она не развивается. Между суставами, например локтевыми, коленными, но уж во всяком случае между пальчиками мы нередко находим тонкие полупрозрачные кожные перепонки, наподобие плавательных мембран у водоплавающих птиц. Кроме недоразвития мускулатуры и подкожного жира имеется еще крайняя степень дряблости связочного аппарата, так что отдельные длинные тонкие членики конечностей чрезмерно пассивно подвижны. Самостоятельные движения детей, страдающих арахнодактилией в тяжелой форме, почти вовсе не бывают или они сведены к минимуму движений некоторых групп мышц. Это крайне ограниченные по объему, замедленные, какие-то неживые, можно сказать, нечеловеческие повороты головы из стороны в сторону. Поэтому эти больные несчастные дети лежат пластом, прикованы к кровати и целиком зависят от обслуживающего персонала. Из-за недоразвития костей, мышц, мезенхимальных образований и жира общий вес тела больных арахнодактилией очень мал. Поразительна общая слабость, и даже в самых благоприятно протекающих случаях у выживающих наступает утомляемость даже после незначительных физических усилий. Таким образом, налицо всегда обязательная и резко выраженная недостаточность статических и механических функций опорно-двигательного аппарата в целом.
При осмотре больного арахнодактилией производит неизгладимое впечатление еще внешний облик личика ребенка. Голова большая, череп увеличен и вытянут в длину, имеется долихоцефалия. Постоянным признаком, вероятно, обусловленным неподвижным лежанием на спинке, является уплощение затылочной области, а также выступление вперед лобных бугров, лоб всегда высокий, крутой. Корень носа широк и погружен. Авторы указывают на изменения челюстей: то это выпячивание нижней челюсти вперед, прогнатия, то это, наоборот, срезанный подбородочек, что придает личику что-то напоминающее птицу. Вообще выражение лица у детей с арахнодактилией не детское, а старческое, скованное, грустное, меланхолическое.
Очень важны для общей клинической характеристики арахнодактилии постоянно выраженные многочисленные и разнообразные неправильности развития глаз. Глаза широко расставлены (гипертелоризм) и лежат глубоко под нависшими над ними выдающимися надбровными дугами. В 30—50% всех случаев арахнодактилии имеется двусторонний симметричный подвывих или вывих хрусталика, смещенного именно кверху. Передняя камера глаза плоска, имеется близорукость. Сетчатка пигментирована. Зрачки узки и при движениях глаз появляется мелкое дрожание радужек (иридодонез). Вывихнутые хрусталики часто мутнеют, что еще в большей степени ухудшает зрение, и обычно с общей неподвижностью тщедушного тельца гармонирует неподвижный невидящий взгляд больного.
Еще чаще, чем поражения глаз, у арахнодактиликов наблюдается неправильное развитие ушных раковин. Они очень велики, не содержат хрящей, а потому мягки, дряблы, лапчаты, легко подвертываются под головку или щечку при боковых движениях. Волосики на голове скудны, сухи и жестки.
Часто, почти в половине всех случаев, определяется и та или иная неправильность, развития сердца, главным образом мышцы его, но нередко и клапанов, перегородки
между камерами, отхождения крупных сосудов, т. е. врожденный порок сердца. В последние годы хорошо рентгенологически изучены [особенно при помощи контрастных методов исследования Стайнбергом (Steinberg)] весьма характерные для арахнодакти-
лии аневризматические расширения аортальных бульбарных синусов, расположенных непосредственно над аортальным артериальным кольцом, подчас огромных уродливых размеров.
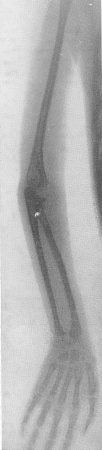
Ри с. 321. Арахнодактилия. Рентгенограмма верхней конечности у 4-летней девочки с резко выраженной клинико-рентгенологической картиной.
Центральная нервная система всегда при арахнодактилии изменена, имеется резкая общая отсталость умственного развития. Явных эндокринных нарушений не бывает. В частности, много внимания уделено исследователями в литературе, да и нами в наших собственных наблюдениях, гипофизарной системе. Но убедительных данных в пользу поражения гипофиза, в особенности его передней доли, мы не получили. И в многочисленных случаях вскрытий, опубликованных в литературе, передняя доля гипофиза, как и все исследованные звенья эндокринной цепи, неизменно оказывались без четких патологических изменений. Определенных биохимических сдвигов крови при арахнодактилйи никто еще не нашел. В частности, изменений кальция и фосфора сыворотки крови, несмотря на всеобщее поражение костной системы, мы ни разу не обнаружили.
Этиология арахнодактилии неизвестна. Поэтому мы ограничиваемся лишь общим неконкретным замечанием о том, что арахнодактилия — это какой-то врожденный глубокий порок развития по преимуществу мезодермальной системы (глаза-то ведь эктодермального происхождения!), какая-то глубинная врожденная мезенхимальная дистрофия.
В последние годы вырабатывается, повидимому, более точное представление о том, что в основе болезни лежит генерализованное поражение эластической арматуры во всем организме, специфическое недоразвитие эластической ткани, где бы она ни располагалась, своеобразный врожденный системный эластоз. Этот порок вызывается пока еще неизвестной нам причиной уже в очень ранней стадии эмбрионального развития.
Распознавание арахнодактилии при условии достаточного знакомства с этой болезненной нозологической формой не представляет никаких трудностей. Рентгенологическое исследование вносит дополнительно к клиническим данным много ценных подробностей, обогащающих наши знания об этой болезни.
Ведущим рентгенологическим признаком арахнодактилии является диспропорция в форме каждой исследованной трубчатой кости, как большой, так и малой (см. рисй 261, Е). До крайности, каррикатурно, как в кривом (цилиндрическом) зеркале, преувеличены длинные размеры и преуменьшены поперечники костей (рис. 321). Арахнодактилия — это диаметральная противоположность хондродистрофии. Кости грацильны, удлинены и истончены, и как уже сказано, это особенно касается фаланг, пястных и плюсневых костей. Большинство авторов при этом указывает на не только относительное, но и абсолютное удлинение конечностей, т. е. истинное увеличение роста тела в длину, которое усиливается с течением времени у тех больных, которые выживают. В школьном возрасте, например, рост больных опережает таковой ихровесников, служащих для контроля, на ряд лет. Интересно, что при крайнем удлинении и истончении костей они не искривляются. Удивительно также, что арахнодактилии совершенно не свойственны патологические переломы, и мы их можем видеть только по вине неосторожного медицинского персонала.
Общий структурный рисунок костей остается довольно правильным. Имеется лишь системная атрофия костей с остеопорозом, рарефикация всего костного вещества, чему, очевидно, в немалой степени способствует общая обездвиженность больных. Корковый слой тонок, а в губчатом веществе трабекулярная сеть редка, трабекул мало, а отдельные трабекулы истончены. Это полностью подтверждается многочисленными гистологическими исследованиями. Мышечные бугры и возвышения, как и эпифизарные концы костей, повсеместно выражены слабо. Наблюдаются псевдоэпифизы.
Темпы окостенения могут быть в единичных случаях извращены, обычно ускорены.
В клинико-рентгенологической картине скелета при арахнодактилии большую роль для формирования общего вида больного имеют еще многочисленные, по существу вторичные деформации. Сюда относятся всегда резко выраженные плоские стопы, нередко конские стопы и тому подобные деформативные процессы, genua valga и recurvata, coxa valga. Слабость мягких тканей суставов, даже без увеличения мышечной тяги, влечет за собой разболтанность больших и малых суставов, подвывихи и резкие вывихи с порочными положениями конечностей. Так, например, мы наблюдали вывихи в суставах больших пальцев стоп, в плечевых и в тазобедренных суставах. В одном нашем случае был асимметричный односторонний вывих бедра, в результате которого бедро легло рядом с краем туловища и колено очутилось в подмышечной впадине! Подчас вызывает удивление крайне подвижная и смещенная далеко в сторону надколенная чашечка.
Грудная клетка обычно резко деформирована, она чаще всего приобретает форму так называемого паралитического или воронкообразного thorax с вдавленной наподобие корыта грудиной. Мы видели однажды весьма странную картину резко сплющенной спереди назад и раздавшейся в стороны, на редкость плоской и широкой, как бы пропущенной через ролики вальца, грудной клетки.
Деформация может еще больше осложниться, если присоединяются те или иные искривления позвоночника — чаще всего сколиозы или ки-фосколиозы, причем отдельные позвонки никаких специальных изменений не представляют. Обезображивается, естественно, и таз, также сплющенный спереди назад.
Арахнодактилию практически ни с какой другой болезнью смешать нельзя, и дифференциальной диагностики ее и не существует, так как в выраженных случаях все клинические и рентгенологические проявления этой болезни сами по себе слишком показательны. Ошибки могут возникнуть только в случае слишком необоснованного расширительного толкования понятия арахнодактилии, т. е. слишком вольной диагностики арахнодактилии у истощенных, крайне худых хронических больных высокого роста.
Спор решает наличие, помимо костных изменений, перечисленных выше, характерных симптомов вовлечения в общий процесс других систем и органов в их оригинальном и неповторяемом сочетании. Вряд ли также возникнет в практической жизни надобность в проведении отличительного распознавания между арахнодактилией и гигантизмом любого другого происхождения, например гипофизарного, скажем, при акромегалии, так как, кроме общего признака удлинения конечностей, здесь больше ничего сходного нет.
И по отношению к арахнодактилии приходится сделать вынужденное признание, что лечить эту болезнь мы в настоящее время в корне не умеем.
Фенилкетонурия ![]()
Московская Медицинская академия им. И.М.Сеченова
Кафедра медицинской генетики
Реферат
Фенилкетонурия
Выполнила: студентка 4 курса
лечебного факультета
33 группы Алиева С.М.
Москва
2004
ФЕНИЛКЕТОНУРИЯ - тяжелое наследственное заболевание, которое
характеризуется главным образом поражением нервной системы.
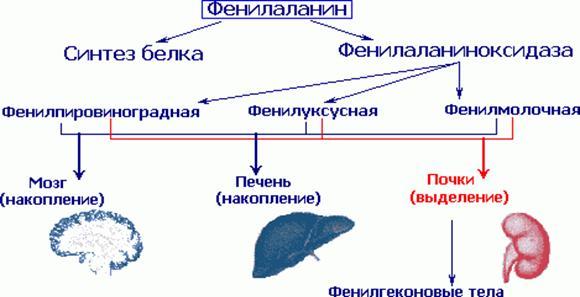
Этиология и патогенез. В результате мутации гена, контролирующего
синтез фенилаланингидроксилазы, развивается метаболический блок на этапе
превращения фенилаланина в тирозин, вследствие чего основным путем
преобразования фенилаланина становится дезаминирование и синтез токсических
производных - фенилпировиноградной, фенил-молочной и фенилуксусной кислот. В
крови и тканях значительно увеличивается содержание фвнилаланина (до 0,2 г/л и
более при норме 0,01-0,02 г/л). Существенную роль в патогенезе болезни играет
недостаточный синтез тирозина, который является предшественником катехоламинов
и меланина. Заболевание наследуется по аутосомно-рецессивному типу.
Фенилкетонурия (ФКУ) - тяжелое наследственное заболевание, наступающее
вследствие врожденного дефекта фермента, отвечающего в организме человека за
нормальный обмен фенилаланина (одной из незаменимых аминокислот, входящих в
состав белка).
При заболевании нарушаются обменные процессы, особенно важные для
развивающегося мозга ребенка. В крови и других жидкостях организма
накапливается в большом количестве фенилаланин и повышено образуются такие
вещества как фенилпировиноградная, фенилмолочная и фенилуксусная кислоты,
которые выделяются в повышенных количествах с мочой. Следствием нарушенного
обмена в мозге является тяжелое психическое недоразвитие. Если не предпринято
своевременное лечение, то больные на всю жизнь остаются глубокими инвалидами.
Поступающий в организм фенилаланин идет на построение белковой цепи или
превращается в тирозин. Отсутствие в печени фермента фенилаланингидроксидазы
препятствует нормальному превращению фенилаланина пищи в тирозин. Поэтому
фенилаланин используется лишь при синтезе белка, а избыток накапливается в
клетках печени и попадает в кровоток, где количество фенилаланина является
токсичным для клеток мозга. Почки не справляются с его реабсорбцией, в
результате чего он выводится с мочой. Именно наличие этого фенилкетона в моче
дало основание назвать соответствующее патологическое состояние
фенилкетонурией.
Варианты ФКУ
Фенилкетонурия 1.
Классическая фенилкетонурия (ФКУ) описана А.Folling.,1934г.
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена,
локализующегося в длинном плече 12 хромосомы.
В основе болезни лежит дефицит фермента фенилаланин-4-гидроксилазы,
обеспечивающего превращение фенилаланина в тирозин. В результате
метаболического блока происходит значительное накопление в тканях и жидкостях
больного организма фенилаланина и таких его производных, как
фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин,
фенилацетилглютамин, и др.
В патогенезе ФКУ имеют значение следующие механизмы:
- Прямое токсическое действие на ЦНС фенилаланина и его производных;
- Нарушение в обмене белков, липо- и гликопротеидов;
- Расстройства транспорта аминокислот;
- Нарушение метаболизма гормонов;
- Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина);
- Нарушение функции печени - диспротеинемия, генерализованная
гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение
окислительной и белковосинтезирующей функции клеточных органелл.
Частота классической ФКУ среди новорожденных по данным массового скрининга в
среднем колеблется от 1:5000 до 1:10000 по разным регионам России.
Фенилкетонурия 2.
Впервые атипичная ФКУ описана I.Smith, 1974г. Заболевание связано с дефицитом
дигидроптеридинредуктазы.
Заболевание наследуется аутосомно-рецессивно. Генный дефект локализуется в
коротком плече 4 хромосомы, участке 4р 15.3.
В результате недостаточности дигидроптеридинредуктазы нарушается
восстановление активной формы тетрагидробиоптерина, участвующего в качестве
кофактора в гидроксилировании фенилаланина, тирозина, и триптофана.
Частота заболевания составляет 1:100000 новорожденных.
Рано начатое лечение способствует нормализации фенилаланина в крови, однако
не предупреждает появление клинической симптоматики, которая развивается в
начале второго полугодия жизни. Фенилкетонурию 2 называют диеторезистентной
ФКУ.
Фенилкетонурия 3.
Этот вариант болезни описал S. Kaufman в 1978 г. Заболевание наследуется
аутосомно-рецессивно и связано с недостаточностью 6-пирувоилтетрагидроптерин
синтетазы, участвующей в процессе синтеза тетрагидробиоптерина. Развивающиеся
при этом расстройства сходны с нарушениями, наблюдаемыми при ФКУ 2.
Частота болезни составляет 1:30000 новорожденных. Фенилкетонурия 3 также
диеторезистентна.
Другие варианты ФКУ:
Эти формы ФКУ связаны с нарушением альтернативных путей обмена фенилаланина.
Формируется метилминдальная ацидурия и парагидроскифенилуксусная ацидурия.
· Материнская фенилкетонурия.
Заболевание развивается у потомков женщин, страдающих ФКУ и не получающих
диету в зрелом возрасте. Патогенез мало изучен, предполагается, что он сходен
с патогенезом остальных форм ФКУ. Тяжесть поражения плода коррелирует с
уровнем фенилаланина в плазме матери. Так как эмбрион особенно чувствителен к
тератогенным воздействиям , рекомендуется начинать диету еще до наступления
беременности. В суточном рационе использовать менее 15-20 мг/кг фенилаланина.
При это важно избегать дефицита незаменимых аминокислот.
· Клиническая фенилкетонурия.
При рождении больные фенилкетонурией не отличаются от других новорожденных.
Манифестация ФКУ происходит обычно в возрасте 2-6 месяцев.
Клинические проявления
Дети с фенилкетонурией (ФКУ) рождаются без каких-либо признаков болезни.
Однако уже на втором месяце можно заметить некоторые физические признаки:
посветление волос, радужек глаз, что особенно заметно у детей, родившихся с
темными волосами. Многие дети очень быстро и чрезмерно прибавляют в весе,
однако остаются рыхлыми, вялыми. У большинства из них рано зарастает большой
родничек. Чаще всего явные признаки болезни обнаруживаются на 4-6 месяце
жизни, когда дети перестают реагировать радостью на обращение к ним,
перестают узнавать мать, не фиксируют взгляд и не реагируют на яркие игрушки,
не переворачиваются на живот, не сидят. В течение многих лет соответствующим
диагностическим тестом служит реакция между фенилпировиноградной кислотой,
которая выделяется с мочой ребенка, и хлорным железом. При положительной
реакции появляется типичное зеленое окрашивание. Кроме того, образуются и
выводятся с мочой другие аномальные метаболиты, такие как фенилмолочная и
фенилуксусная кислоты. Последнее соединение «пахнет мышами», так что болезнь
легко диагностировать по запаху; именно так она и была впервые обнаружена.
По мере прогрессирования болезни могут наблюдаться эпилептиформные приступы -
развернутые судорожные и бессудорожные типа кивков, поклонов, вздрагиваний,
кратковременных отключений сознания. Гипертония отдельных групп мышц
проявляется своеобразной "позой портного" (поджатые ноги и согнутые руки).
Могут наблюдаться гиперкинезы, атаксия, тремор рук, иногда парезы по
центральному типу. Дети нередко белокурые со светлой кожей и голубыми
глазами, у них часто отмечаются экзема, дерматиты. Обнаруживается склонность
к артериальной гипотензии.
Первыми проявлениями болезни служат:
- вялость ребенка, отсутствие интереса к окружающему;
- повышенная раздражительность, беспокойство;
- срыгивание, рвота;
- судорожные эквиваленты: спонтанный рефлекс Моро, спонтанный рефлекс
Бабинского, сосательные автоматизмы, приапизм у мальчиков, атетозные
движения;
- судорожный синдром;
- заплесневелый, мышиный, волчий запах мочи и пота.
При отсутствии лечения формируется задержка статико-моторного и психоречевого
развития, умственная отсталость достигает, как правило, глубокой степени
(идиотия или имбецильность, глубокая психическая инвалидность).
Дигностика фенилкетонурии
Чрезвычайно важно установить диагноз в доклинической стадии или по крайней
мере не позднее 2-го месяца жизни, когда могут проявиться первые признаки
болезни. Для этого всех новорожденных обследуют по специальным профаммам
скрининга, выявляющего повышение концентрации фенилаланина в крови уже в
первые недели жизни. Оптимальные сроки обследования новорожденных - 5-14-день
жизни . Каждого ребенка, у которого обнаруживаются признаки задержки развития
или минимальная неврологическая симптоматика, необходимо обследовать на
патологию обмена фенилаланина. Используют микробиологический и
флюорометрический методы определения концентрации фенилаланина в крови, а
также пробу Фелинга на фенилпировиноградную кислоту в моче (прибавление
нескольких капель 5% раствора треххлористого железа и уксусной кислоты к моче
больного приводит к появлению зеленой окраски пятна на пеленке). Эти и другие
подобные методы относятся к категории ориентировочных, поэтому при
положительных результатах требуется специальное обследование с использована
ем точных количественных методов определения содержания фенилаланина в крови
ц моче (хроматография аминокислот, использование аминоанализаторов и др.),
которое осуществляется централизованными биохимическими лабораториями.
Дети требуют специального наблюдения и лечения в медико-генетических центрах
(кабинетах поликлиник).
Дифференциальный диагноз проводят с внутричерепной родовой травмой,
внутриутробными инфекциями.
ФКУ может быть диагностирована на основе обнаружения следующих признаков:
o стойкой гиперфенилаланинемии (более 240 ммоль/л);
o вторичного дефицита тирозина;
o экскреции фенилкетонов с мочой (проба Феллинга на экскрецию
фенилпировиноградной кислоты).
В настоящее время, согласно приказу Минздрава России № 316 от 30.12.93
проведение неонатального скрининга на ФКУ стало обязательным. Скрининирующие
тесты должны быть простыми, недорогими и информативными.
Этим требованиям отвечают методы, используемые для ранней диагностики ФКУ:
o микробиологический тест Гатри;
o метод флюоресцирующих антител (лабораторный комплекс "Флюороскан",
позволяющий проводить 800 проб в час);
o метод тонкослойной хроматографии.
Оптимальные сроки обследования новорожденных - доношенных, зрелых - 5-6 день
жизни; недоношенных, незрелых, больных - 10-14 день жизни.
Трактовка результатов:
1 группа.
Уровень фенилаланина не превышает 200 ммоль/л (1-3 мг%) - норма;
2 группа.
Уровень фенилаланина составляет 200-500 ммоль/л (3-10 мг%) -
гиперфенилаланинемия. В эту группу входят дети с транзиторной
гиперфенилаланинемией, вследствие незрелости ферментных систем печени и
больные ФКУ. За данной группой проводится наблюдение по следующему плану:
если в течение 6 недель при еженедельном исследовании уровень фенилаланина
остается менее 500 ммоль/л, контроль за уровнем фенилаланина крови проводят
до 1 года первоначально каждые 3 месяца, а затем каждые 6 мес. жизни. При
уровне более 500 ммоль/л назначается диетотерапия.
3 группа.
Уровень фенилаланина превышает 500 ммоль/л (более 10 мг%), диагностируется
ФКУ и с момента постановки диагноза назначается диетотерапия.
В настоящее время разрабатываются и внедряются молекулярно-генетические
методы диагностики генного дефекта при ФКУ. Прямая диагностика мутантного
гена проводится с помощью синтетических олигонуклеотидных зондов, этот метод
пригоден для дородовой диагностики ФКУ и выявления гетерозиготного
носительства.
Помимо молекулярно-генетического анализа, выявление гетерозигот может
осуществляться биохимическими тестами после нагрузки фенилаланином в дозе 25
мг/кг.
Диеторезистентные формы ФКУ диагностируют при помощи:
o исследования биоптеринов мочи ;
o перорального нагрузочного теста с тетрагидробоиптерином (через 4-6
часов после однократной дачи нагрузки в дозе 7,5 мг/кг массы тела происходит
резкое снижение и нормализация уровня фенилаланина в крови с одновременным
повышением уровня тирозина);
o исследования активности дигидроптеридинредуктазы и 6-
пирувоилтетрагидроптерин синтетазы в культуре кожных фибробластов,
эритроцитах, гепатоцитах.
Наследственность
Болезнь наследуется по рецессивному типу: т.е. болеют сестры и братья из
одной семьи, а родители здоровы, хотя и являются гетерозиготными носителями
гена ФКУ. Ген фенилкетонурии встречается в среднем у 1-2 на 100 человек, но
болезнь может возникнуть лишь в том случае, если и мать и отец ребенка
являются носителями этого гена, и ребенок унаследует его в двойном наборе.
Поэтому болезнь встречается значительно реже, чем распространен ген. Больные
ФКУ (обладатели двух патологических генов) могут иметь детей с
фенилкетонурией только при вступлении в брак с носителями таких же генов. При
вступлении в брак с лицами свободными от гена ФКУ, дети не болеют этим
заболеванием.
Лечение фенилкутонурии и прогноз
Если ничего не предпринимать, фенилкетонурия приводит к весьма тяжелым
последствиям - развивается олигофрения. К счастью, этот трагический исход
можно предотвратить, если поставить правильный диагноз при рождении. В наши
дни это легко осуществимо в ходе массового обследования (скрининга)
новорожденных с целью выявления ФКУ.
Главным способом лечения является диетотерапия, ограничивающая поступление в
организм фенилаланина; приступить к ней нужно немедленно после
установления диагноза. При ранней диагностике это гарантирует нормальное
нервно-психическое развитие ребенка.
Диетотерапия, как единственный эффективный метод лечения ФКУ, должна
применятся с первых месяцев жизни ребенка, тогда поражение мозга не
разовьется. Важно ограничить количество потребляемого фенилаланина таким
образом, чтобы обеспечить его поступление в организм в количествах,
необходимых и достаточных для роста и развития, но предотвратив его
накопление в жидкостях тела. Кроме диетотерапии необходим постоянный
медицинский контроль за умственным и физическим развитием ребенка.
Очень важно! Применение диетотерапии на позднем этапе не вернет ребенку
нормального интеллекта. Дети, у которых это заболевание не диагностируют
сразу при рождении, а выявляют по умственной отсталости, не могут быть
излечены.
По достижении 12-14 лет такие дети могут переходить на нормальное питание и
никаких признаков отравления фенилаланином у них не наблюдается. Однако
женщина, которая в детстве переболела ФКУ, должна снова перейти на диету и
употреблять только продукты с пониженным содержанием фенилаланина перед
зачатием, и оставаться на этой диете во время беременности и кормления
грудью. Если она не сделает этого, то ее ребенок подвергается риску
замедленного физического и умственного развития, даже если его отец не
является носителем гена ФКУ.
Диетотерапия
Единственным лечением, способным предотвратить развитие слабоумия или
уменьшить его степень, является диета, исключающая поступление в организм
фенилаланина сверх того минимального количества, которое необходимо для
образования собственных белков организма и его роста. Поэтому смысл
диетического лечения сводится к резкому ограничению естественного белка с
пищей.
Для такого ограничения приходится полностью исключить из питания ребенка
такие богатые белками продукты как мясо, колбасы, рыбу, бульоны, яйца,
творог, сыр, мучные изделия, каши из естественных круп, фасоль, орехи,
шоколад. Меню для детей составляется из фруктов, овощей, крахмальных изделий,
жиров, со строгим учетом содержания в них фенилаланина.
Назначают белковые гидролизаты (цимогран, лефанолак, берлофен, гипофенат) или
аминокислотные смеси, лишенные фенилаланина, которые становятся главными
продуктами питания, обеспечивающими потребность в белке: "Лофенолак",
"Фенилфри" (США), "Берлофен", "Апонти", "Гипофенат" у детей до 4-5 лет и
"Нофелан" - у детей старше 5 лет. Белковые гидролизаты вводят с фруктовыми и
овощными соками, пюре, супами.
Лечение проводят под контролем содержания фенилаланина в крови, добиваясь
поддержания его уровня в пределах 0,03-0,04 г/л.
Строгое ограничение белков животного происхождения требуется на протяжении
первых 2-3 лет жизни. Наиболее рационально отменять диетическое лечение в
возрасте 7-8 лет.
-Препараты с промедиаторным действием:
1.Наком (комбинация карбиДОФА и левоДОФА) - доза 100-375 мг/сутки в течение
3-4 недель, перерыв между курсами 1,5-2 месяца;
2.Лево-дофа - доза 10-15 мг/кг в сутки;
3.5-окситриптофан - доза 10 мг/кг сутки.
-Ноотропные препараты
-Витамины
-ЛФК, массаж
При диеторезистентных формах лечение включает назначение тетрагидробиоптерина
- доза 10-20 мг/кг в сутки.
Профилактика фенилкетонурии
1. Выявление гетерозиготных носителей. Большое значение имеет
специальное наблюдение за семьями риска, т. е. за такими семьями, где уже
имелись дети с фенилкетонурией. Новорожденные из этих семей должны быть
подвергнуты обязательному биохимическому исследованию и при показаниях к
раннему лечению.
2. Внедрение программ массового скрининга новорожденных для раннего
выявления ФКУ и своевременного назначения диетотерапии. Выявление и лечение
детей по программам массового скрининга также позволяет предупредить развитие
тяжелой психической инвалидности.
3. Пренатальная диагностика: Предложен ДНК-зонд для пренатальной
диагностики фенилкетонурии в семьях высокого риска.
Болезнь Вильсона-Коновалова
ЗАДАТЬ
ВОПРОС ДОКТОРУ
Поделиться…
|Версия для печати|
Болезнь Вильсона-Коновалова (или
гепатоцеребральная дистрофия) - редкое
наследственное заболевание, в основе
которого лежит генетически обусловленное
нарушение обмена меди с избыточным ее
накоплением преимущественно в печени
и нервной
системе.
Этиология и патогенез.
В основе лежит аутосомно-рецессивное наследственное нарушение метаболизма меди; ген расположен в длинной части хромосомы 13. Распространенность в различных регионах мира в среднем 1:30000 при частоте гетерозиготного носительства около 1 %. Первоначально ген экспрессируется в печени, почках, плаценте. Продукт гена представляет собой катионтранспортирующий Р-тип АТФазного протеина. Следствием генетического дефекта является различной степени выраженности нарушение функции внутриклеточного транспорта меди. Это ведет к снижению экскреции меди с желчью и накоплению её в гепатоцитах. С пищей в сутки поступает 2-5 мг меди. Она всасывается в кишечнике, поступает в печень, где связывается с синтезируемым печенью церулоплазмином, циркулирует в сыворотке крови, избирательно захватывается органами и экскретируется с желчью. В норме экскреция меди с желчью 2 мг в сутки, при болезни Вильсона-Коновалова - только 0.2-0.4 мг, что приводит к повышенному накоплению меди в организме. Включение меди в церулоплазмин происходит в аппарате Гольджи при участии гена гепатоцеребральной дистрофии. Незначительная часть меди находится в крови в ионизированной форме в виде лабильного комплекса с альбумином и выделяется с мочой. При болезни Вильсона-Коновалова увеличена абсорбция меди в кишечнике, снижена экскреция меди с желчью. Снижение экскреции меди связано с дефектом гена гепатоцеребральной дистрофии, определяющего транспорт меди в аппарат Гольджи и последующее выделение лизосомами в желчь. Нарушается процесс включения меди в церулоплазмин. Из-за недостаточного использования меди происходит её депонирование в печени, мозге, почках, роговице. Депонированная в печени медь вторично ингибирует синтез церулоплазмина. Уровень церулоплазмина в сыворотке крови имеет диагностическое, но не патогенетическое значение. У 5 % больных определяется нормальный уровень церулоплазмина. При биопсии печени у таких больных имеется избыточное количество меди, также увеличивается содержание меди в крови и тканях, выделение её с мочой. Медь, являясь прооксидантом, оказывает токсическое действие на организм. Её накопление ведет к повышенной продукции свободных гидроксильных радикалов. При обследовании больных болезнью Вильсона-Коновалова и животных с экспериментальной перегрузкой медью в плазме крови определяется снижение уровня витамина Е, увеличение циркулирующих продуктов перекисного окисления липидов; в печени снижены уровни восстановленного глутатиона и витамина Е. Митохондрии печени являются мишенями действия оксидантов. Нарушение дыхательной цепи и снижение активности цитохром-С-оксидазы увеличивает продукцию свободных радикалов благодаря утечке электронов из дыхательной цепи. Свободная медь, накапливаясь в тканях, блокирует SH-группы ферментов, участвующих в окислительно-восстановительных реакциях. Это приводит к энергетическому голоданию, к которому наиболее чувствительна ЦНС. В начале заболевания, когда клинические признаки отсутствуют (I стадия), медь накапливается в цитозоле печеночных клеток. Медь, связанная с SH-группами цитозольных протеинов, затрудняет секрецию гепатоцитами белков и триглицеридов. Наступает стеатоз гепатоцитов и появление телец Маллори. Во II стадии медь перераспределяется из цитозоля в лизосомы гепатоцитов. Часть поступает в кровь. В связи с низкой специфической активностью лизосом билиарная экскреция меди понижается. Медь вызывает переокисление липидов и повреждение лизосомальных мембран с последующим выходом вредных кислых гидролаз в цитоплазму. Наблюдаются некроз гепатоцитов, развитие хронического гепатита и гемолитической анемии. В III стадии усиленное накопление меди в печени приводит к фиброзу и циррозу. Повышенное накопление меди в головном мозге, роговице, дистальных отделах почечных канальцев приводит к развернутой картине болезни.
Морфология
В печеночной ткани наблюдаются жировая дистрофия гепатоцитов, перипортальный фиброз, субмассивные некрозы гепатоцитов, макронодуллярный цирроз. В почках - жировая и гидропическая дистрофия с отложением меди в проксимальных канальцах.
Клиническая картина
Клинические
проявления:
1. Печеночные -
цирроз печени, хронический активный
гепатит, фульминантная печеночная
недостаточность.
На начальной стадии
изменения в печени неспецифические -
мелко- и среднекапельная жировая
дистрофия, некрозы единичных гепатоцитов,
перипортальный фиброз. Далее развивается
клиника хронического гепатита высокой
степени активности с желтухой, высоким
уровнем аминотрансфераз,
гипергаммаглобулинемией. При
прогрессировании - цирроз печени с
портальной гипертензией и печеночноклеточной
недостаточностью.
Фульминантная
печеночная недостаточность - редкое
проявление гепатоцеребральной дистрофии.
Развивается у подростков и молодых
людей. Характерные признаки, позволяющие
дифференцировать её с фульминантным
гепатитом вирусной этиологии: небольшое
повышение активности трансаминаз (с
преобладанием повышения активности
АСТ), низкий уровень щелочной фосфатазы,
крайне низкий уровень альбумина в
сыворотке крови, высокий уровень прямого
и непрямого билирубина (внутрисосудистый
гемолиз), гемоглобинурия, высокий уровень
меди в сыворотке печени и её экскреции
с мочой. Часто сопровождается гемолитической
анемией, связанной с массивным
высвобождением меди из печени. Единственный
эффективный метод лечения - трансплантация
печени.
Существует абдоминальная
форма Керара - поражение печени преобладает
на всем протяжении болезни и рано
осложняется печеночной недостаточностью.
В дебюте - развитие отечно-асцитического
синдрома, степень выраженности которого
не соответствует выраженности других
признаков портальной гипертензии.
Постоянное наличие большого количества
несвязанной меди в сыворотке крови и
повышенное отложение её не только в
печени, но и в других органах приводит
к повреждению головного мозга, роговицы,
почек, скелета, гемолизу эритроцитов.
2.
Неврологические -
экстрапирамидные, церебеллярные,
псевдобульбарные нарушения, судорожные
припадки.
Две основные формы заболевания
- ригидно-аритмогиперкинетическая, или
ранняя, и дрожательная - значительно
различаются по своим клиническим
проявлениям. Первая характеризуется
быстрым развитием общей ригидности и
наличием неритмичных гиперкинезов
атетоидного или торсионно-спастического
характера. Ригидность распространяется
на мышцы туловища, конечностей и на
мышцы, участвующие в глотании и речевом
акте. Отмечаются амимия, дисфагия,
дизартрия. Походка становится скованной,
подпрыгивающей. Ригидность мышц может
приступообразно усиливаться, особенно
в связи с произвольными движениями и
под влиянием эмоций. Больные часто
застывают в самых неудобных позах. В
дистальных отделах конечностей нередко
образуются контрактуры. Нарастающая
ригидность быстро приводит к полной
обездвиженности. Эта форма заболевания
начинается в детском возрасте - от 7 до
15 лет. Висцеральные расстройства могут
проявляться раньше - в возрасте 3-5 лет.
Как правило, выражены признаки печеночной
патологии, которые часто предшествуют
развитию неврологической
симптоматики.
Флексорно-экстензорный
тремор. Его выраженность колеблется от
едва заметного дрожания рук до тремора
всего тела. Тремор усиливается при
волнении и целенаправленных действиях.
Умеренный тремор у ряда больных может
иметь акцент на одной стороне. Тремор
пальцев вытянутых рук типичный,
"порхающий".
Мышечная дистония
наблюдается у всех больных. Проявления
дрожательно-ригидной формы различной
выраженности. Определяется гипомимия,
гиперсаливация, затрудненная монотонная
речь, снижение интеллекта. Акинетико-ригидная
форма сопровождается ярко выраженным
ригидным синдромом, затрагивающим
различные группы мышц. В развернутой
стадии выявляется гиперкинез по типу
"бьющихся крыльев", к которому
могут присоединяться интенционный
компонент, дизартрия, дисфагия, мозжечковые
расстройства, миоклонии. Без специфической
терапии нарастание симптоматики приводит
к выраженным контрактурам, обездвиженности,
грубой деменции.
У больных с
экстрапирамидной патологией могут
развиваться пирамидные моно- и гемипарезы.
Такие случаи относятся к
экстрапирамидно-корковой форме
гепатоцеребральной дистрофии, которая
отличается от других форм значительным
поражением коры больших полушарий. У
больных часто отмечаются эпилептические
припадки общего и особенно джексоновского
характера, тяжелое нарушение интеллекта
с грубыми нарушениям личности. Психические
нарушения могут иметь место и у больных
с другими формами заболевания. Они
характеризуются изменениями
эмоционально-волевой сферы, снижением
психической активности и интеллекта.
Наряду с этим наблюдаются случаи
доброкачественного течения
гепатоцеребральной дистрофии, когда у
больных в течение длительного времени
неврологическая симптоматика отсутствует
или имеются очень легкие симптомы,
которые не нарушают их трудоспособности.
Такие больные, как правило, выявляются
случайно при обследовании семей больных,
с развернутой картиной заболевания.
3.
Психиатрические -
нарушения в эмоциональной сфере, психоз,
нарушения поведения, познавательной
деятельности.
4. Гематологические -
гемолиз, анемия, тромбоцитопения,
нарушения свертывающей системы крови.
У 15 % больных могут наблюдаться явления
острого внутрисосудистого гемолиза.
Гемолиз обычно временный, проходит
самостоятельно, предшествуя ярким
клиническим признакам поражения печени
в течение нескольких лет. Иногда может
протекать одновременно с острой
печеночной недостаточностью. Предполагается
влияние больших количеств свободной
меди в плазме на мембраны эритроцитов
и гемоглобин.
5. Почечные -
канальцевые нарушения (частичный или
полный синдром Фанкони), снижение
клубочковой фильтрации, нефролитиаз.
Поражение
почек проявляется периферическими
отеками, микрогематурией, незначительной
протеинурией, повышением концентрации
креатинина сыворотки крови. Как ранний
симптом может наблюдаться макро- и
микрогематурия. Наиболее часто в моче
обнаруживают треонин, тирозин, лизин,
валин, фенилаланин.
6. Офтальмологические -
кольцо Кайзера-Флейшера, катаракта
(содержащие медь отложения в капсуле
хрусталика).
7. Эндокринологические -
аменорея, спонтанные аборты, задержка
полового развития, гинекомастия,
гирсутизм, ожирение, гипопаратироидизм.
8.
Сердечно-сосудистые -
кардиомиопатия, аритмия.
9.
Мышечно-скелетные -
остеомаляция, остеопороз, артропатия,
артралгии.
10. Желудочно-кишечные -
холелитиаз, панкреатит, спонтанный
бактериальный перитонит.
11.
Дерматологические -
голубые лунки у ногтевого ложа, сосудистая
пурпура, гиперпигментация кожи, acantosis
nigricans.
Диагностика
Подозрение на наличие болезни Вильсона-Коновалова должно возникнуть при:
неуточненной этиологии хронического гепатита и цирроза;
фульминантной печеночной недостаточности;
необъяснимом повышении уровня аминотрансфераз;
наличии соответствующих неврологических изменений неустановленной этиологии, изменении поведения;
психических симптомах, сочетающихся с признаками заболевания печени;
необъяснимой приобретенной гемолитической анемии;
семейном анамнезе по гепатоцеребральной дистрофии.
Основные (скрининговые) тесты для диагностики болезни Вильсона-Коновалова:
обнаружение кольца Кайзера-Флейшнера: не обнаруживается у 50-62 % больных без неврологических симптомов; может отсутствовать у 5 % больных с начальными признаками поражения ЦНС;
выявление снижения содержания церулоплазмина в сыворотке крови до уровня < 20 мг/дл (норма 25-50 мг/дл): уровень < 5 мг/дл - абсолютное доказательство болезни Вильсона-Коновалова. Умеренное снижение может встречаться у гетерозиготных носителей гена, при циррозе печени другой этиологии, при синдроме мальабсорбции, нефротическом синдроме и др.; у 10-15 % больных с абдоминальной формой заболевания уровень церулоплазмина может быть в пределах нормы;
увеличение содержания не связанной с церулоплазмином меди в сыворотке крови (300 мкг/л и >);
повышение содержания меди в органах, в частности в биоптатах печени (свыше 250 мкг/г сухой массы);
повышение экскреции меди с мочой (более 200 мкг в сутки при норме < 70 мкг в сутки);
D-пеницилламиновый тест - повышение суточной экскреции меди до уровня >1500 мкг, в норме значительного увеличения экскреции меди с мочой не наблюдается;
высокий уровень включения изотопа меди в церулоплазмин - в норме - отсутствие пика включения через 48 часов; тест диагностически значим только у больных с нормальным уровнем церулоплазмина;
генетические исследования: значимы у сибсов и других членов семьи пробанда.
Множество мутаций гена ATP7B, ответственного за развитие болезни Вильсона-Коновалова, не позволяет использовать молекулярно-генетические методы для скрининговой диагностики заболевания, но они могут применяться для постановки диагноза у сибсов и членов семьи пробанда. Все сибсы пробанда подлежат обязательному обследованию для исключения болезни Вильсона, риск наличия которой для них составляет 25%. Возможны сложности дифференциальной диагностики бессимптомных гомозиготных и гетерозиготных носителей гена ATP7B, так как у 10-20 % могут наблюдаться снижение уровня церулоплазмина и отклонения от нормы в обмене меди, а при бессимптомном течении заболевания возможны отсутствие кольца Кайзера-Флейшера и гиперкупрурии. Для количественного определения меди в биоптатах печени используют спектрофотометрию, рентгеноструктурный анализ. Также для диагностики используют поглощение печенью радиоактивной меди. Соотношение радиоактивности печени через 24 и 2 часа после внутривенного введения радионуклида меди в норме равно 1.4-9, а при болезни Вильсона-Коновалова 0.2-0.3. Гетерозиготные носители и больные с другими заболеваниями печени имеют соотношение, равное единице. Кинетика радиоактивной меди позволяет дифференцировать болезнь Вильсона-Коновалова от гепатоцеребрального синдрома при заболеваниях печени.
Лечение
Терапия направлена на выведение избытка меди из организма для предупреждения её токсического воздействия. Назначают диету № 5, богатую белком, с ограничением содержащих медь продуктов (баранина, куры, утки, колбасы, рыба, ракообразные, шампиньоны, кресс-салат, щавель, лук-порей, редис, бобовые, орехи, чернослив, каштаны, шоколад, какао, мед, перец и др.). Основа терапии - использование препаратов, связывающих медь и выводящих её из организма:
Британский антилюизит (2,3-димеркаптопропанол) - вводят внутримышечно по 1.25-2.5 мг/кг 2 раза в день в течение 10-20 дней, перерыв между курсами 20 дней. Другая методика применения: введение 200-300 мг 2 раза в день в течение нескольких месяцев до получения эффекта. Применение препарата ограничено из-за болезненности инъекций и появления признаков интоксикации при длительном лечении.
Унитиол 5% - по 5-10 мл ежедневно или через день, на курс 25-30 внутримышечных инъекций. Повторные курсы через 2-3 месяца.
D-пеницилламин. Увеличивает выведение меди с мочой: образует комплексы, которые легко фильтруются через почечные клубочки. Дозы от 0.3-1.3 до 3-4 граммов в сутки в зависимости от величины экскреции меди с мочой. Оптимальная доза препарата 0.9-1.2 грамма в сутки.
Осложнения терапии D-пеницилламином:
Гематологические - злокачественный агранулоцитоз, преходящая тромбо- и лейкопения часто наблюдаются в первые 6 недель лечения, контрольные анализы крови делают сначала 3 раза в неделю, потом 1 раз в месяц.
Почечные - нефротический синдром выявляется обычно в период от 2 месяцев до 2 лет после начала лечения. Он может исчезать спонтанно или при применении глюкокортикоидов.
Кожные - локальная или генерализованная эритема, уртикарные высыпания, геморрагические кожные высыпания.
Аллергические - для устранения используют преднизолон.
При выраженных токсических эффектах применяется другое хелатообразующее соединение - триэтилентетрамин. При непереносимости обоих препаратов - тетратиомолибдат. Он блокирует всасывание меди в кишечнике, а также связывает медь в тканях в метаболически инертную форму. Альтернативный метод лечения - применение значительно менее токсичного, чем D-пеницилламин, сульфата цинка. Он тормозит абсорбцию меди в кишечнике. Дозировка - 200 мг 3 раза в день за 30 минут до еды. Также применяют витамины В1 и В6, так как избыточное количество меди блокирует их активность. Показаны препараты, улучшающие обмен гепатоцитов, антиоксиданты. Доза препарата должна устанавливаться ежегодно, а при длительном лечении каждые 2 года на основании выделения меди с мочой, контрольных биопсий печени и определения содержания меди в биоптатах печени. Клиническое улучшение под влиянием лечения выражается в сглаживании неврологической симптоматики, снижении активности воспалительного процесса в печени. При успешной терапии D-пеницилламином выведение меди с мочой увеличивается в 3-5 раз. В первые 2-3 недели от начала лечения может наблюдаться усиление неврологической симптоматики и ухудшение функционального состояния печени, которое затем сменяется улучшением, обычно через несколько недель или месяцев. Есть описания полного исчезновения активности хронического гепатита и цирроза по данным биопсии печени спустя годы после применения препарата.
Трансплантация печени
Показана при фульминантной печеночной недостаточности, прогрессировании печеночной недостаточности на фоне хронического гепатита и цирроза печени при неэффективности медикаментозной терапии.
Прогноз
Течение заболевания прогрессирующее, ведущее к инвалидизации. Прогноз улучшается при назначении адекватной терапии на ранних стадиях заболевания. Терапия на поздней стадии существенно не влияет на развитие осложнений. Смерть наступает преимущественно в молодом возрасте, как правило, от осложнений цирроза печени (кровотечения из варикознорасширенных вен пищевода, печеночная недостаточность) или фульминантного гепатита, реже - от осложнений, связанных с поражением ЦНС.
Профилактика
Ранняя диагностика заболевания. При выявлении дефектного гена в гомозиготном состоянии лечение медьхелатирующими препаратами может быть начато в раннем детском возрасте.
Беременность
Своевременная терапия болезни Вильсона-Коновалова приводит к полному восстановлению функции печени, исчезновению симптомов, сохранению трудоспособности больных, значительному увеличению выживаемости, а также восстановлению репродуктивной функции. Данные наблюдения беременных, страдающих болезнью Вильсона-Коновалова, свидетельствуют о том, что беременность ими хорошо переносится. Это связано с тем, что плод забирает часть меди у матери, а также, возможно, с повышением уровня церулоплазмина во время беременности. Поражение печени при болезни Вильсона-Коновалова, в том числе на стадии цирроза в отсутствие выраженных признаков портальной гипертензии, не является показанием к прерыванию беременности. Основную проблему в ведении таких больных представляет необходимость продолжения медьэлиминирующей терапии на протяжении беременности, так как полная ее отмена может привести к опасному для жизни женщины обострению заболевания. В то же время отрицательные эффекты на развитие плода (тератогенные, нарушение развития соединительной ткани) при применении D-пеницилламина полностью не исключаются. Рекомендуется снижение дозы D-пеницилламина в 3-4 раза в период беременности. В последние годы для поддержания ремиссии заболевания у беременных предлагается применение препаратов цинка. Несмотря на пока небольшой опыт лечения этими препаратами во время беременности, предполагается безопасность их применения для плода.
Литература:
С.Д. Подымова. Болезни печени: Руководство. - 4 издание, переработанное и дополненное. - М.: ОАО "Издательство "Медицина", 2005. - 768 с. (сс. 567-578).
Ш. Шерлок, Дж. Дули. Заболевания печени и желчных путей: Практическое руководство.: Перевод с английского./Под редакцией З.Г. Апросиной, Н.А. Мухина. - М.: Гэотар Медицина, 1999. - 864 с. (сс. 476-483).
Справочник практического врача по гастроэнтерологии./Под редакцией В.Т. Ивашкина, С.И. Рапопорта. - М.: Советский спорт, 1999. - 432 с. (сс. 175-177).
Справочник Харрисона по внутренним болезням./Под редакцией К. Иссельбахера, Е. Браунвальда, Дж. Вильсон и др. - СПб: Издательство "Питер", 1999. - 976 с. (сс. 786-787).
Т.М. Игнатова. Ранняя диагностика болезни Вильсона-Коновалова: радикальное улучшение прогноза. Врач, 2004, № 12, сс. 36-39.
Болезнь Тея-Сакса (первая часть)
Болезнь Тея-Сакса (БТС) (также известная как GM2 ганглиолипидоз, дефицит гексозаминидазы или ранняя детская амавротическая идиотия) - это аутосомно-рецессивное генетическое заболевание, которое вызывает прогрессирующее ухудшение умственных и физических способностей ребенка. Первые признаки заболевания, обычно проявляются в возрасте примерно 6 месяцев. Расстройство, как правило, приводит к смерти больного человека в возрасте около 4 лет.
Заболевание вызвано генетическим дефектом конкретного гена. Если ребенок, поражен БТС, то это означает, что он унаследовал по одной копии дефектного гена от каждого родителя. Заболевание проявляется тогда, когда в нервных клетках мозга накапливается опасное количество ганглиозидов, что в результате приводит к преждевременной смерти этих клеток. На сегодня не существует никаких эффективных лекарств или иных методов лечения для этой болезни. БТС - встречается довольно редко, по сравнению с другими рецессивными заболеваниями, такими как, например, муковисцидоз (кистозный фиброз) и серповидно-клеточная анемия - которые более распространенные.
Болезнь названа в честь британского офтальмолога Уоррена Тея (который первым описал красное пятно на сетчатке глаза в 1881 году) и американского невролога Бернарда Сакса, который работал в больнице Маунт Синай в Нью-Йорке (он описал клеточные изменения, происходящие при БТС и в 1887 году отметил, увеличение частоты заболевания среди евреев ашкенази, которые этнически походят с территории Восточной Европы).
Исследования заболевания, которые проводились в конце ХХ века показали, что болезнь Тея-Сакса обусловленамутациями гена HEXA, который находится на 15 хромосоме. На сегодня уже выявлено большое количество мутаций HEXA, а новые исследования дают информацию о новых мутациях. Эти мутации очень часто встречаются в нескольких популяций. Количество носителей среди франко-канадцев (живущих на юго-востоке провинции Квебек) почти такое, как среди евреев-ашкенази, однако, мутации, вызывающие БТС среди этих этнических групп - разные. Многие представители этнической группы Кейджн (которые сегодня живут на территории южной Луизианы) являются носителями таких же мутаций, которые наиболее распространены среди евреев-ашкенази. Как уже было сказано, эти мутации очень редки и не встречаются среди генетически изолированных популяций. То есть, болезнь может возникнуть лишь от наследования двух независимых мутаций в гене HEXA.
Классификация и симптомы
Болезнь Тея-Сакса классифицируется по разным формам, в зависимости от времени возникновения неврологических симптомов. Форма заболевания отражает вариант мутации.
Детская форма болезни Тея-Сакса.
На протяжении первых шести месяцев после рождения дети развиваются нормально. Но, после того как нервные клетки накапливают ганглиозиды и, таким образом растягиваются, наблюдается непрерывное ухудшение умственных и физических способностей больного. Ребенок становится слепой, глухой, и не может глотать. Мышцы начинают атрофироваться, вследствие чего наступает паралич. Смерть обычно наступает в возрасте до четырех лет.
Подростковая форма болезни Тея-Сакса.
Эта форма заболевания встречается крайне редко и обычно проявляет себя у детей в возрасте от 2 до 10 лет. У них развиваются когнитивно-моторные проблемы, проблемы с речью (дизартрия), глотанием (дисфагия), шаткость походки (атаксия), возникает спастичность. Пациенты с подростковой формой БТС обычно умирают в возрасте от 5 до 15 лет.
Взрослая форма болезни Тея-Сакса (англ. LOTS).
Редкая форма расстройства, известная как взрослая форма болезни Тея-Сакса или поздняя форма болезни Тея-Сакса (LOTS), возникает у пациентов в возрасте от 20 до 30 лет. LOTS часто неправильно диагностируется, и, как правило, не имеет летального исхода. Она характеризуется нарушением походки и прогрессирующим ухудшением неврологических функций. Симптомами данной формы, которая возникает в подростковом или раннем взрослом возрасте, являются: проблемы с речью и глотанием, шаткость походки, спастичность, снижение когнитивных навыков, возникновения психических заболеваний, в частности шизофрении в виде психоза.
Еще до 1970-х и 80-х годов, когда стала известна молекулярная природа заболевания, взрослую и подростковую формы почти никогда не рассматривали, как формы болезни Тея-Сакса. БТС, которая возникала в подростковом или взрослом возрасте часто диагностировали как другие неврологические расстройства, например как атаксию Фридрейха. Люди, пораженные БТС во взрослом возрасте, часто передвигаются с помощью инвалидного кресла, однако многие из них живут почти полноценной жизнью, однако лишь в том случае, если приспособятся к физическим и психиатрическим осложнениям (которые можно контролировать с помощью медицинских препаратов).
Журналистка Джанет Сильвер Гент (Janet Silver Ghent) описала опыт девушки Веры, которая была родом из русско-еврейской семьи, иммигрировавшей в США, когда она была еще ребенком. Двадцать лет назад, когда Вере Песотчинской (Vera Pesotchinsky's) было 14, у нее появились трудности с речью (ее речь стала нечеткой, невнятной), именно поэтому родители обратились за помощью к логопеду. Позже, у девушки начали возникать проблемы с координацией, иногда она даже падала, кроме того, Вера не могла четко сделать определенные координированные движения (например, не могла почистить картошку). Мать Веры обратилась за консультацией к специалистам в области неврологии и психиатрии. И только через 12 лет и после большого количества неправильных диагнозов, у девушки, наконец, диагностировали LOTS. Несмотря на свою инвалидность, Вера окончила колледж Уэлсли (Wellesley College) и получила научную степень магистра управления (англ. МВА) в Университете Санта-Клара (Santa Clara University). Согласно словам Дж. Гент Вера живет самостоятельно, ежедневно работает в семейном бизнесе, и кроме того, она твердо убеждена, что не стала жертвой БТС, а ее пример указывает на то, что с этим заболеванием можно вполне нормально жить. Вера является примером для всех больных БТС, она обращается ко всем пораженным этой болезнью людей и добавляет мотивации для борьбы за здоровье: «Конечно, вы можете «развалиться» на части и быть больным, но вы можете и лечиться. Делайте все что вы можете для лечения болезни, потому что если бы я этого не делала, то моя ситуация была бы значительно хуже ».
Патофизиология
Болезнь Тея-Сакса возникает вследствие недостаточной активности фермента гексозаминидазы А, который катализирует биодеградацию определенного класса жирных кислот известных как ганглиозиды. Гексозаминидаза А является жизненно необходимым гидролитическим ферментом, который находится в лизосомах и разрушает липиды. Когда гексозаминидаза А перестает функционировать должным образом, липиды накапливаются в головном мозге и препятствуют нормальным биологическим процессам. Ганглиозиды производятся и биодеградируют быстро, в самом начале жизни, в то время как развивается мозг. Пациенты и носители болезни Тея-Сакса, могут быть определены путем сдачи относительно простого биохимического анализа крови, который определяет активность гексозаминидазы А.
Для гидролиза GM2-ганглиозида необходимыми являются три белка. Два из них - субъединицы гексозаминидазы А, а третий - небольшой гликолипидный транспортный белок, GM2 белок-активатор (GM2A), выступающий в качестве субстрата для конкретного кофактора фермента. Дефицит любого из этих белков приводит к накоплению ганглиозид, главным образом в лизосомах нервных клеток. Болезнь Тея-Сакса (вместе с GM2 ганглиозидозом и болезнью Сандхоффа) возникает через генетические мутации, унаследованные от обоих родителей, которые отключают или тормозят процесс расщепления этих веществ. Большинство мутаций БТС, как считают ученые, не влияет на функциональные элементы белка. Вместо этого, они вызывают неправильное накопления или хранения фермента, в связи с чем, внутриклеточная транспортировка становится невозможной.
Генетика
БТС - это аутосомно-рецессивное генетическое заболевание. Это означает, что в том случае, если оба родителя являются носителями дефектного гена, то риск того, что новорожденный ребенок будет больным составляет 25%. Аутосомные гены - это хромосомные гены, они не находятся на одной из половых хромосом. Каждый человек является носителем двух копий каждого аутосомно гена, по одной унаследованной от каждого из родителей. Если оба родителя - носители мутации, то согласно генетических законов Менделя, вероятность передачи болезни ребенку составляет 25%. Как и все генетические заболевания, ХТС может возникнуть в любом поколении, при этом не важно, когда впервые возникла мутация. Хотя мутации, которые вызывают БТС, встречаются довольно редко.
Аутосомно-рецессивные заболевания возникают, если ребенок наследует две копии дефектного аутосомно гена, то есть когда одна копия не может участвовать в процессе транскрипции или экспрессии в качестве функционального продукта для образования фермента.
БТС вызвана мутацией гена HEXA, который находится на 15 хромосоме и кодирует деятельность альфа-субъединицы лизосомного фермента бета-N-ацетилгексоаминидазы А. К 2000 году, было определено более 100 мутаций в гене HEXA, однако, еще и сегодня количество известных мутаций постоянно увеличивается. Эти мутации происходят в виде вставок пар оснований, их делеций, это могут быть сплайс-сайт мутации, точечные мутации и другие. Каждая из этих мутаций изменяет белковый продукт, и тем самым подавляет активность фермента. Проведенные недавно демографические исследования, показали, какие из мутаций возникают и распространяются в пределах малочисленных этнических групп. Основой для исследования стали следующие группы:
• Евреи Ашкенази. Для них характерна вставка четырех пар оснований в 11 экзоне (1278insTATC). Это приводит к повреждению рамки считывания для HEXA гена. Эта мутация является самой распространенной среди евреев ашкенази и приводит к появлению инфантильной формы болезни Тея-Сакса.
• Кейджн. Эта этническая группа (население которой проживает сегодня в Южной Луизиане, США), была отделена от остального населения на несколько веков через языковые различия. БТС вызывают те же мутации, которые являются самыми распространенными среди евреев Ашкенази. Исследователи изучили родословную всех носителей из нескольких семей Луизианы и определили семейную пару, у которых впервые родился ребенок с БТС. Однако, это супруги не были потомками евреев, живших во Франции в XVIII веке.
• Франко-Канадцы. Для этой популяции характерна делеция длинной последовательности нуклеотидов, которая влечет возникновение тех же патологий, которые вызывают вышеописанные мутации (встречающиеся у евреев ашкенази и кейджн). Как и количество евреев ашкенази, так и количество лиц франко-канадского населения быстро увеличилось, из небольшой группы учредителей, однако, при этом они оставались изолированными от остального населения через географические, культурные и языковые барьеры. Ранее считалось, что мутации в этих двух группах населения - идентичны, а распространенность БТС в восточной провинции Квебека вызвана потоком генов. Некоторые ученые тогда утверждали, что «сексуально активный еврейский предок» привел к возникновению болезнетворной мутации среди франко-канадского населения. Эта теория в узких кругах (среди ученых генетиков) стала известна под названием «Гипотеза о еврейском торговце мехами». Однако последующие исследования показали, что две мутации не имеют между собой ничего общего.
В 1960-х и начале 1970-х годов, когда впервые стали известны биохимические основы болезни Тея-Сакса, последовательность мутации, которая вызвала любое генетическое заболевание не могла быть точно определена. Исследователи той эпохи еще не знали на сколько распространенным может быть полиморфизм. Знание того времени отражает именно «гипотеза о еврейском торговце мехами», ведь согласно ей, возможно распространение только одной мутации между популяциями. Дальнейшие исследования показали, что БТС может вызвать большое количество мутаций, каждая из которых вызывает появление различных форм заболевания. Именно БТС стала первым генетическим нарушением, которое показало возможность возникновения такого явления как компаундная (объединенная) гетерозиготность. Такие основательные знания стали доступны благодаря тому, что БТС была первой болезнью, для определения которой начали широко использовать генетический скрининг.
Само явление компаудной гетерозиготности объясняет различные формы расстройства, в том числе и возникновение взрослой формы БТС. Потенциально, заболевание может возникнуть в результате наследования двух отличных мутировавших копий гена HEXA, по одной от каждого родителя. Классическая инфантильная форма БТС возникает, если ребенок унаследовал от обоих родителей одинаково мутировавшие копии гена, нарушение функции которого вызывает полную инактивацию процесса расщепления (биодеградации) ганглиозидов. Взрослая форма БТС возникает через наследование различных мутаций, и несмотря на то, что человек может быть гетерозиготным, он может унаследовать две различные мутации гена HEXA, совместное действие которых приводит к инактивации, изменению или уменьшению активности нужного фермента. Если у пациента хотя бы одна копия гена HEXA позволяет гексозаминидазе осуществлять свои функции, то результатом этого является возникновение взрослой формы БТС.
У гетерозиготных носителей, т.е. у лиц, которые унаследовали лишь одну мутантную аллель, уровень активности фермента тоже несколько понижен, однако никаких признаков или симптомов заболевания у них не проявляется.Брюс Корф объясняет, почему у носителей рецессивных мутаций, как правило, не возникает симптомов генетического заболевания: «биохимические основы доминирования аллелей дикого типа над мутантными аллелями при врожденных метаболических заболеваниях, можно понять, изучив процесс функционирования белков. Ферменты - белки, которые катализируют химические реакции, т.е. для нормального осуществления реакции катализа необходимо лишь небольшое количество вещества. Если у гомозиготных особей, ген кодирующий деятельность фермента мутирован, то это приводит к уменьшению активности фермента или вообще к его отсутствию в организме, т.е. у этого человека будет проявляться ненормальный фенотип. Зато у гетерозиготных лиц уровень активности фермента составляет не менее 50% от нормального уровня, через экспрессию (действие) аллелей «дикого типа». Обычно этого достаточно, чтобы предотвратить нарушение фенотипа».
Диагностика
Совершенствование разработанных методов тестирования позволило невропатологам гораздо точнее диагностировать болезнь Тея-Сакса и другие неврологические заболевания. Однако, иногда болезнь Тея-Сакса диагностируется неправильно, потому что врачи не уверены в том, это разновидность генетического заболевания, характерного для евреев ашкенази.
Пациенты с данным заболеванием имеют "вишневое" пятно макулы, которое легко выявить врачу с помощью офтальмоскопа на сетчатке глаза. Это пятно является участком сетчатки, которая увеличивается через накопления ганглиозидов в окружающих ганглиозных клетках сетчатки (они являются нейронами центральной нервной системы). Таким образом, только вишневая пятна макулы является той частью сетчатки, которая обеспечивает нормальное зрение. Микроскопический анализ нейронов показывает, что эти клетки растянуты (нагруженные ганглиозидами) в связи с избыточным накоплением ганглиозидов. Без использования молекулярных методов диагностики, только вишневое пятно макулы является характерной чертой и признаком при диагностировании всех ганглиозидозов.
В отличие от некоторых других лизосомальных болезней накопления (например, болезни Гоше, Нимана-Пика,Сандхоффа), гепатоспленомегалия не является характерным признаком болезни Тея-Сакса.
Журналист Аманда Пазорник (Pazornik) описывает опыт семьи Арбогаст: "Пейтон была красивой девочкой, - но она не могла сидеть, переворачиваться, играть со своими игрушками. Причем симптомы Пейтон прогрессивно ухудшались. Громкий, непонятный шум пугал ее. Неспособность координировать движения мышц рта и языка приводила к тому, что она могла поперхнуться во время приема пищи и была причиной чрезмерного выделения слюны». Поскольку ни один из родителей Пейтон не был евреем, ее врачи не подозревали, что она поражена болезнью Тея-Сакса, до тех пор, пока ей не исполнилось 10 месяцев, именно тогда офтальмолог заметил вишневое пятно макулы в ее глазах. Пейтон умерла в 2006 году в возрасте 3,5 лет. Это характерный ход заболевания. Ребенок становится все более "ленивым" из-за нарушения нейродегенеративного развития и показывает чрезмерный рефлекс от гиперакузии. Больной человек становится все более вялым и имеет проблемы при потреблении пищи. Могут стать заметными спастичность и двигательные расстройства. Это расстройство является наиболее распространенным среди евреев Ашкенази.