

Основные стехиометрические законы химии( закон сохранения массы, закон Авогадро, закон постоянства состава, закон кратных и объемных отношений.)
Стехиометрия — раздел химии, в котором рассматриваются массовые или объемные соотношения между реагирующими веществами.
1. Закон сохранения массы веществ
Масса веществ, вступивших в реакцию, равна массе веществ, образовавшихся в результате реакции.
С точки зрения атомно-молекулярного учения этот закон объясняется тем, что при химических реакциях общее количество атомов не изменяется, а происходит лишь их перегруппировка. Закон сохранения массы веществ является основным законом химии, все расчёты по химическим реакциям производятся на его основе. Именно с открытием этого закона связывают возникновением современной химии как точной науки.
С законом сохранения массы веществ тесно связан закон сохранения энергии:
энергия не возникает из ничего и не исчезает бесследно, но одни её виды могут превращаться в другие в строго эквивалентных количествах.
Так при разложении воды, кислот, щелочей или солей посредством электрического тока электрическая энергия превращается в химическую. То же наблюдается при зарядке аккумулятора. Обратный процесс — превращение химической энергии в электрическую — происходит при разрядке аккумулятора.
Альберт Эйнштейн показал, что между массой тела и его энергией Е существует связь, выражаемая соотношением:
E = mc2
где с - скорость света в вакууме, равная 300 000 км/с. Это уравнение применимо ко всем энергетическим процессам; в том числе к химическим и ядерным реакциям. Из него следует, что если масса системы изменяется, то происходит изменение и её энергии, и наоборот: изменение внутренней энергии системы всегда сопровождается изменением массы. Вследствие химических реакций всегда выделяется или поглощается энергия. Поэтому строго говоря, масса веществ, участвующих в этих реакциях, должна изменяться: при выделении теплоты — уменьшаться, а при поглощении — увеличиваться. Однако вследствие очень большой величины множителя с2изменения массы при химических реакциях настолько малы, что определить их существующими методами невозможно.
Рассчитаем, например, какому количеству энергии соответствует изменение массы на 1 г:
Е = 1·10-3·(3·108)2= 9·1013Дж.
При образовании 1 моля воды из газообразных водорода и кислорода выделяется 285,9 кДж энергии, то уменьшение массы составит 3,2·10-9г. Эта величина далеко за пределами возможностей взвешивания. Тепловые эффекты химических реакций таковы по своей величине, что изменение массы веществ в их результате не могут быть изменены. Поэтому закон сохранения массы веществ не могут быть измерены. Поэтому закон сохранения массы веществ соблюдается практически при всех химических реакциях.
2. Закон постоянства состава
Закон сохранения массы послужил основой для изучения количественного состава различных химических соединений. Многочисленные опыты показали, что качественный и количественный состав различных сложных веществ постоянен и не зависит от способа их получения.
Так, 44 г углекислого газа (СО2) соединяясь с 56 негашеной извести (СаО), образуют 100 г мела (СаСО3). При этом никакие другие вещества не образуются, а данные соединения вступают в реакцию полностью. Если через негашёную известь пропустить избыток углекислого газа, то он не будет реагировать с образовавшимся мелом.
Чистая (без примесей) вода независимо от того, получена она синтезом из водорода и кислорода, при нейтрализации щёлочи кислотой или при любой другой реакции, и чистая природная вода всегда состоит из водорода и кислорода в соотношении 1:8 по массе.
Французский учёный Жан Луи Пруст, обобщив большой экспериментальный материал о составе различных веществ, сформулировал в 1799 г закон постоянства состава:
каждое химическое соединение имеет постоянный качественный и количественный состав независимо от способа его получения.
Этот закон находится в полном соответствии с атомно-молекулярным учением. Действительно, молекула любого вещества состоит из вполне определённого количества атомов, имеющих постоянную массу. Поэтому её массовый состав и, следовательно, массовый состав вещества постоянны независимо от способа его получения. Такие соединения называются дальтониды.
Современная химия располагает данными, из которых следует, что закону постоянства состава подчиняются главным образом вещества, имеющие молекулярную структуру, если же вещества не имеют молекулярной структуры, то возможны отклонения от этого закона.
Действительно соединения переменного состава, называемые бертолиды, существуют и с каждым годом их открывают всё больше. Эти соединения не имеют определённой химической формулы. Впервые бертолиды были обнаружены в системах, состоящих их нескольких металлов (интерметаллические сплавы), затем среди оксидов, сульфидов, селенидов металлов и др. Например, оксид титана (II) имеет состав от ТiO0,59до TiO1,33, в соединении таллия с висмутом на 1 часть таллия приходится от 1,24 до 1,82 частей висмута по массе. В природе бертолиды распространены значительно шире, чем дальтониды.
Отклонения от закона постоянства состава может быть обусловлено не только изменениями атомного состава соединений, но и причинами связанными с наличием в природе изотопов. Например, для водорода известны три изотопа с массовыми числами 1 (протий), 2 (дейтерий) и 3 (тритий). Естественно, что в молекуле воды, образованные первым, вторым или третьим изотопом, на 1 атом кислорода приходятся 2 атома водорода (атомный состав постоянен), однако процентное содержание кислорода в этих соединениях переменно и составляет соответственно 88,89; 80 и 72,73 %.
3. Закон кратных отношений. Закон объёмных отношений.
Известны случаи, когда два элемента, соединяясь между собой в различных количественных соотношениях, образуют несколько химических соединений. Так, углерод с кислородом образуют два соединения следующего состава: монооксид углерода (угарный газ) СО — 3 весовых части углерода и 4 весовых части кислорода; диоксид углерода (углекислый газ) СО2— 3 весовых части углерода и 8 весовых частей кислорода.
Количество весовых частей кислорода, приходящееся в этих соединениях на одно и то же количество углерода (3 весовых части), соотносится между собой как 4:8 или 1:2.
Азот с кислородом образует пять оксидов (табл. 1).
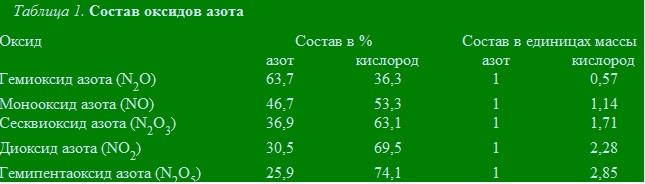
Количество весовых частей кислорода приходящееся в этих соединениях на одну весовую часть азота соотносится между собой как 0,57 : 1,14 : 1,71 : 2,28 : 2,85 = 1 : 2 : 3 : 4 : 5.
Данные о количественном составе различных соединений, образованных двумя элементами, и исходя из атомистических представлений, английский химик Джон Дальтон в 1803 году сформулировал закон кратных отношений:
если два элемента образуют между собой несколько соединений, то на одно и то же весовое количество одного элемента приходятся такие весовые количества другого элемента, которые относятся между собой как небольшие целые числа.
То, что элементы вступают в соединения определенными порциями, явилось ещё одним подтверждением правильности атомистического учения и объяснения с его позиций химических процессов.
Однако атомистические представления сами по себе не могли объяснить, например, количественных соотношений, которые наблюдаются в химических реакциях между газами.
Французский учёный Ж. Гей-Люссак, изучая химические реакции между газообразными веществами, обратил внимание на соотношения объёмов реагирующих газов и газообразных продуктов реакции. Он установил, что 1 л хлора целиком вступает в реакцию с 1 л водорода с образованием 2 л хлороводорода; 1 л кислорода взаимодействует без остатка с 2 л водорода, образуя 2 л водяного пара. Эти опытные данные Гей-Люссак обобщил в законе объёмный отношений:
Объёмы реагирующих газообразных веществ относятся между собой и к объемам образующихся газообразных веществ как небольшие целые числа.
4. Закон Авогадро
Итальянский физик Амедео Авогадро сделал очень важное дополнение к атомистической теории. Он ввел понятие о молекуле как мельчайшей частице вещества, способной к самостоятельному существованию. Он использовал понятие молекулы для объяснения простых объёмных отношений между реагирующими газами. В 1811 году он выдвинул следующую гипотезу:
в равных объёмах различных газов при одинаковых условиях (давлении и температуре) содержится равное число молекул.
Авогадро принял, что молекулы простых газов состоят из двух атомов: О2, Н2, Cl2, N2. При этом допущении реакцию между хлором и водородом, приводящую к образованию хлороводорода можно представить уравнением:
Н2+ Сl2= 2 HСl
из которого видно, что из одной молекулы водорода и одной молекулы хлора образуются две молекулы хлороводорода. Следовательно, и объём, занимаемый хлороводородом, должен быть вдвое больше объёма вступившего в реакцию водорода или хлора. Суммарный же объём исходных газов в соответствии с приведённым уравнением должен быть равен объёму образовавшегося хлороводорода.
Гипотеза Авогадро была подтверждена большим числом экспериментальных данных и вошла в науку под названием закона Авогадро. Этот закон вводил в науку представление о молекулах, как мельчайших частицах элемента.
2) Квантово-механическая модель строения атома. Корпускулярно- волновые свойства электрона. Уравнение Планки, Луи Де Бройля, Шредингера. Волновая функция, электронное облако, электронная орбиталь.
Квантово-механическая модель атома
Современная модель атома является развитием планетарной модели. Согласно этой модели, ядро атома состоит из положительно заряженных протонов и не имеющих заряда нейтронов и окружено отрицательно заряженными электронами. Однако представления квантовой механики не позволяют считать, что электроны движутся вокруг ядра по сколько-нибудь определённым траекториям (неопределённость координаты электрона в атоме может быть сравнима с размерами самого атома).
Химические свойства атомов определяются конфигурацией электронной оболочки и описываются квантовой механикой. Положение атома в таблице Менделеева определяется электрическим зарядом его ядра (то есть количеством протонов), в то время как количество нейтронов принципиально не влияет на химические свойства; при этом нейтронов в ядре, как правило, больше, чем протонов (см.: атомное ядро). Если атом находится в нейтральном состоянии, то количество электронов в нём равно количеству протонов. Основная масса атома сосредоточена в ядре, а массовая доля электронов в общей массе атома незначительна (несколько сотых процента массы ядра).
Массу атома принято измерять в атомных единицах массы, равных 1⁄12от массы атома стабильного изотопа углерода12C.
Корпускуля́рно-волново́й дуали́зм— принцип, согласно которому любой объект может проявлять как волновые, так и корпускулярные свойства. Был введён при разработке квантовой механики для интерпретации явлений, наблюдаемых в микромире, с точки зрения классических концепций. Дальнейшим развитием принципа корпускулярно-волнового дуализма стала концепция квантованных полей в квантовой теории поля.
Как классический пример, свет можно трактовать как поток корпускул (фотонов), которые во многих физических эффектах проявляют свойства электромагнитных волн. Свет демонстрирует свойства волны в явлениях дифракции и интерференции при масштабах, сравнимых с длиной световой волны.
3) Электронная структура атомов. Порядок заполнения электронный орбиталей: принцип наименьшего запаса энергии, правила Клечковского, Гунда, Паули. Проиллюстрировать примерами электронных оболочек атома s-,p-,f-,d- семейств.
Квантовые числа используют для описания состояния электрона в атоме. Наряду с квантовыми числами для тех же целей используют:
-диаграммы уровней энергии атома;
-электронные конфигурации.
1. Диаграмма уровней энергии
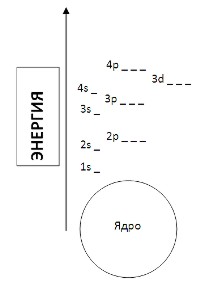
На рисунке показана диаграмма уровней атома, при помощи которой можно описать электроны любого атома.
Заполняя диаграмму, следует придерживаться следующих несложных правил:
сначала электроны заполняют самые низкие из свободных уровней энергии;
если конкретный уровень имеет более одного подуровня (для n=3;4…), то каждый из подуровней будет заполняться только одним электроном до тех пор, пока все подуровни этого уровня не будут иметь по 1 электрону. Только после этого эти подуровни будут заполняться вторыми электронами. (Правило Хунда).
Правило Хундаопределяет порядок заполнения орбиталей определённого подслоя и формулируется следующим образом: суммарное спиновое число электронов данного подслоя должно быть максимальным.
Это означает, что в каждой из орбиталей подслоя заполняется сначала один электрон, а только после исчерпания незаполненных орбиталей на эту орбиталь добавлется второй электрон. При этом на одной орбитали находятся два электрона с полуцелыми спинами противоположного знака, которые спариваются (образуют двухэлектронное облако) и, в результате, суммарный спин орбитали становится равным нулю.
Другая формулировка: Ниже по энергии лежит тот атомный терм, для которого выполняются два условия. 1. Мультиплетность максимальна 2. При совпадении мультиплетностей суммарный орбитальный момент L максимален.
При́нцип Па́ули (принцип запрета)— один из фундаментальных принципов квантовой механики, согласно которому два тождественных фермиона не могут одновременно находиться в одном квантовом состоянии.
В атоме каждый электрон располагается так, чтобы его энергия была минимальной (что отвечает наибольшей связи его с ядром).
Энергия электрона в основном определяется главным квантовым числом n и побочным квантовым числом l, поэтому сначала заполняются те подуровни, для которых сумма значений квантовых чисел n и l является наименьшей. Например, энергия электрона на подуровне 4s меньше, чем на подуровне 3d, так как в первом случае n + 1 = 4 + 0 = 4, а во втором n + l = 3 + 2 =5; на подуровне 5s (n + l = 5 + 0 = 5) энергия меньше, чем на 4d (n + l = 4 + 2 = 6); на 5р (n + l = 5 + 1 = 6) энергия меньше, чем на 4f (n + l = 4 + 3 = 7) и т.д.
Правило Клечковского — эмпирическое правило, описывающее энергетическое распределение орбиталей в многоэлектронных атомах.
Первое и второе правила Клечковского часто не разделяют, а считают одним совместным правилом правила Клечковского: при увеличении заряда ядра атомов заполнение энергетических уровней происходит от орбиталей с меньшим значением суммы главного * и орбитального * квантовых чисел (n+l) к орбиталям с большим значением этой суммы.
Число электронов на внешнем энергетическом уровне электронной оболочки атома равно номеру группы для химических элементов главных подгрупп.
Как ранее было сказано, электрон движется не по орбите, а по орбитали и не имеет траектории.
Пространство вокруг ядра, где наиболее вероятно нахождение данного электрона, называется орбиталью этого электрона, или электронным облаком.
Орбитали, или подуровни, как их еще называют, могут иметь разную форму, и их количество соответствует номеру уровня, но не превышает четырех. Первый энергетический уровень имеет один подуровень (s), второй – два (s,p), третий – три (s,p,d) и т.д. Электроны разных подуровней одного и того же уровня имеют разную форму электронного облака: сферическую (s), гантелеобразную (p) и более сложную конфигурацию (d) и (f). Сферическую атомную орбиталь ученые договорились называть s-орбиталью. Она самая устойчивая и располагается довольно близко к ядру.
Чем больше энергия электрона в атоме, тем быстрее он вращается, тем сильнее вытягивается область его пребывания, и, наконец, превращается в гантелеобразную p-орбиталь:
![]()
Электронное облако такой формы может занимать в атоме три положения вдоль осей координат пространства x, y и z. Это легко объяснимо: ведь все электроны заряжены отрицательно, поэтому электронные облака взаимно отталкиваются и стремятся разместиться как можно дальше друг от друга.
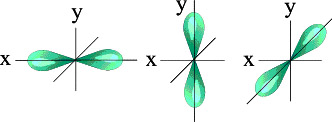
Три p – орбитали
Итак, p-орбиталей может быть три. Энергия их, конечно, одинакова, а расположение в пространстве – разное.
Составить схему последовательного заполнения электронами энергетических уровней
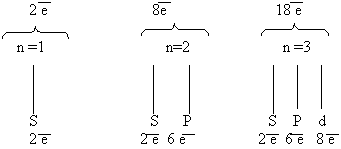
Теперь мы можем составить схему строения электронных оболочек атомов:
Определяем общее число электронов на оболочке по порядковому номеру элемента.
Определяем число энергетических уровней в электронной оболочке. Их число равно номеру периода в таблице Д. И. Менделеева, в котором находится элемент.
Определяем число электронов на каждом энергетическом уровне.
Используя для обозначения уровня арабские цифры и обозначая орбитали буквами s и p, а число электронов данной орбитали арабской цифрой вверху справа над буквой, изображаем строение атомов более полными электронными формулами. Ученые условились обозначать каждую атомную орбиталь квантовой ячейкой – квадратиком на энергетической диаграмме:
На s-подуровне может находиться одна
атомная орбиталь![]()
а на p-подуровне их может быть уже три
–![]()
(в соответствии с тремя осями координат):
Орбиталей d– и f-подуровня в атоме может быть уже пять и семь соответственно:

4) Квантово-механическая модель строения атома. Понятие о волновой функции. Квантовые числа. Написать электронную формулу элемента с порядковым номером 25 и определить значения квантовых чисел для электронов внешнего уровня.
Что такое квантовые числа? Какие значения они могут принимать?
Решение.Движение электрона в атоме носит вероятностный характер. Околоядерное пространство, в котором с наибольшей вероятностью (0,9—0,95) может находиться электрон, называется атомной орбиталью (АО). Атомная орбиталь, как любая геометрическая фигура, характеризуется тремя параметрами (координатами), получившими название квантовых чисел (n,ℓ,mℓ). Квантовые числа принимают не любые, а определенные, дискретные (прерывные) значения. Соседние значения квантовых чисел различаются на единицу. Квантовые числа определяют размер (n), форму (ℓ) и ориентацию (m1) атомной орбитали в пространстве. Занимая ту или иную атомную орбиталь, электрон образует электронное облако, которое у электронов одного и того же атома может иметь различную форму. Формы электронных облаков аналогичны АО. Их также называют электронными или атомными орбиталями. Электронное облако характеризуется четырьмя квантовыми числами (n, ℓ, mℓ и ms). Эти квантовые числа связаны с физическими свойствами электрона, и числоn(главное квантовое число) характеризует энергетический (квантовый) уровень электрона; числоℓ(орбитальное) — момент количества движения (энергетический подуровень), числоmℓ(магнитное) — магнитный момент,ms— спин. Спин электрона возникает за счет вращения его вокруг собственной оси. Электроны в атоме должны отличаться хотя бы одним квантовым числом (принцип Паули), поэтому в АО могут находиться не более двух электронов, отличающихся своими спинами (ms= ± 1/2).
Таблица 1
Значения квантовых чисел и максимальное число электронов на квантовых уровнях и подуровнях
|
Квантовый |
Магнитное квантовое число, ml |
Число квантовых состояний (орбиталей) |
Максимальное число электро-нов | |||||
|
уровень |
подуровень | |||||||
|
обозна-чение |
главное квантовое число, n |
обозна-чение |
орбитальное квантовое число, l |
в под-уровне (2l+1) |
в уров-не n2 |
в под-уровне (2l+1) |
в уров-не n2 | |
|
K |
1 |
s |
0 |
0 |
1 |
1 |
2 |
2 |
|
L |
2 |
s |
0 |
0 |
0 |
4 |
2 |
8 |
|
|
|
p |
1 |
-1; 0; +1 |
3 |
|
6 |
|
|
M |
|
s |
0 |
0 |
1 |
|
2 |
|
|
|
|
p |
1 |
-1; 0; +1; |
3 |
9 |
6 |
18 |
|
|
|
d |
2 |
-2;-1; 0; +1; +2 |
5 |
|
10 |
|
|
N |
4 |
s |
0 |
0 |
1 |
|
2 |
|
|
|
|
p |
1 |
-1; 0; +1 |
3 |
|
6 |
|
|
|
|
d |
2 |
-2; -1; 0; +1; +2 |
5 |
16 |
10 |
32 |
|
|
|
f |
3 |
-3; -2; -1; 0; +1;+2; +3; |
7 |
|
14 |
|
Составьте электронные формулы атомов элементов с порядковыми номерами 16 и 22. Покажите распределение электронов этих атомов по квантовым (энергетическим) ячейкам.
Решение.Электронные формулы отображают распределение электронов в атоме по энергетическим уровням, подуровням (атомным орбиталям). Электронная конфигурация обозначается группами символов, гдеn— главное квантовое число,ℓ— орбитальное квантовое числов (вместо него указывают соответствующее буквенное обозначение — s, p, d, f), х — число электронов в данном подуровне (орбитали). При этом следует учитывать, что электрон занимает тот энергетический подуровень, на котором он обладает наименьшей энергией — меньшая сумма n + ℓ (правило Клечковского). Последовательность заполнения энергетических уровней и подуровней следующая:
1s→2s→ 2р→ 3s→ 3р→ 4s→ 3d→ 4р→ 5s→ 4d→ 5р→ 6s→ 5d1→4f→ 5d→ 6р→ 7s →6d1 →5f→ 6d→ 7р
Так как число электронов в атоме того или иного элемента равно его порядковому номеру в таблице Д.И. Менделеева, то для элементов № 16 (сера) и № 22 (титан) электронные формулы имеют вид
16S 1s2 2s2 2р63s23р4; 22Ті 1s22s22р63s23р63d24s2
Электронная структура атома может быть изображена также в
виде схем размещения электронов в
квантовых (энергетических) ячейках,
которые являются схематическим
изображением атомных орбиталей (АО).
Квантовую ячейку обозначают в виде
прямоугольника,
кружка О или линейки ─, а электроны в
этих ячейках обозначают стрелками ↑↓.
В каждой квантовой ячейке может быть
не более двух электронов с противоположными
спинами. В данном пособии применяют
прямоугольники.
Орбитали данного подуровня заполняются
сначала по одному электрону с одинаковыми
спинами, а затем по второму электрону
с противоположными спинами (правило
Хунда).
атома может быть изображена также в
виде схем размещения электронов в
квантовых (энергетических) ячейках,
которые являются схематическим
изображением атомных орбиталей (АО).
Квантовую ячейку обозначают в виде
прямоугольника,
кружка О или линейки ─, а электроны в
этих ячейках обозначают стрелками ↑↓.
В каждой квантовой ячейке может быть
не более двух электронов с противоположными
спинами. В данном пособии применяют
прямоугольники.
Орбитали данного подуровня заполняются
сначала по одному электрону с одинаковыми
спинами, а затем по второму электрону
с противоположными спинами (правило
Хунда).
Волнова́я фу́нкция, или пси-функция— комплекснозначная функция, используемая в квантовой механике для описания чистого состояния системы. Является коэффициентом разложения вектора состояния по базису (обычно координатному):

Физический смысл волновой функции заключается в том, что согласно копенгагенской интерпретации квантовой механики плотность вероятности нахождения частицы в данной точке пространства в данный момент времени считается равной квадрату абсолютного значения волновой функции этого состояния в координатном представлении.
Волновая функция в различных представлениях
Набор координат, которые выступают в роли аргументов функции, представляет собой полную систему коммутирующих наблюдаемых. В квантовой механике возможно выбрать несколько полных наборов наблюдаемых, поэтому волновая функция одного и того же состояния может быть записана от разных аргументов. Выбранный для записи волновой функции полный набор величин определяет представление волновой функции. Так, возможны координатное представление, импульсное представление, в квантовой теории поля используется вторичное квантование и представление чисел заполнения или представление Фока и др.
Если волновая функция, например, электрона в атоме, задана в координатном представлении, то квадрат модуля волновой функции представляет собой плотность вероятности обнаружить электрон в той или иной точке пространства. Если эта же волновая функция задана в импульсном представлении, то квадрат её модуля представляет собой плотность вероятности обнаружить тот или иной импульс.
5) Периодическая система химических элементов Д.Ю. Менделеева. Построение сисемы с точки зрения строения атома: периоды, группы, подгруппы, s-,p-,f-,d- элементы и их свойства.
Периодическая система элементов (таблица Менделеева) — классификация химических элементов, позволяющая выявить зависимость их различных свойств от числа протонов в атомном ядре. Всего предложено несколько сот вариантов изображения периодической системы (аналитических кривых, таблиц, геометрических фигур и т. п.). В современном варианте системы предполагается сведение элементов в двухмерную таблицу, в которой каждый столбец (число столбцов составляет 8) определяет основные физико-химические свойства, а строки представляют собой периоды, в определенной мере подобные друг другу.,
Таблица Д.И. Менделеева состоит из 8 групп. Физический смысл номера группы заключается в том, что номер группы соответствует валентности входящих в него элементов.
Если элемент обладает переменной валентностью, то номер группы соответствует наивысшей валентности данного элемента.
Каждая группа делится на главную (А) и побочную (В) подгруппу.
Главная подгруппа содержит элементы и малых, и больших периодов (S и p - элементы). Побочные подгруппы включают в себя d и f - элементы, т.е. элементы только больших периодов.
Элементы, составляющие одну подгруппу, имеют близкие химические свойства, т.к. имеют сходные валентные окончания. Химические свойства элементов обуславливаются их валентными окончаниями.
Валентное окончание элемента зависит от его положения в таблице Менделеева.
Все элементы одной подгруппы имеют одинаковую конфигурацию валентных окончаний, но разные квантовые числа.
С увеличением главного квантового числа у элементов данной подгруппы усиливаются металлические свойства, что обусловлено, в первую очередь, увеличением промежуточных электронных слоёв между ядром атома и его валентным слоем.
Элементы, составляющие одну группу, но разные подгруппы, имеют различные валентные окончания и различные химические свойства.
Электронное облакоp-электрона имеет осевую симметрию, поэтому возможны три ориентации p-облаков вдоль трех координатных осей, что соответствует трем p-орбиталям. Поскольку одна орбиталь может вмещать два электрона, на каждом p- подуровне может находиться не больше 6 электронов. На третьем и последующих уровнях помимо s- и p-подуровней имеется еще d-подуровень. вмещающий 10 электронов, располагающихся на пяти d-орбиталях.
На четвертом и последующих уровнях есть помимо перечисленных выше еще и f-подуровни, вмещающий 14 электронов, располагающихся на семи f-орбиталях.
6) Периодический закон и периодическая система химических элементов Д.Ю. Менделеева. Основные характеристики атома (радиус атома, энергия ионизации, энергий сродства к электрону, электроотрицательность.) Изменение этих свойств по периодам и группам.
Периодическая система элементов (таблица Менделеева) — классификация химических элементов, позволяющая выявить зависимость их различных свойств от числа протонов в атомном ядре. Всего предложено несколько сот вариантов изображения периодической системы (аналитических кривых, таблиц, геометрических фигур и т. п.). В современном варианте системы предполагается сведение элементов в двухмерную таблицу, в которой каждый столбец (число столбцов составляет 8) определяет основные физико-химические свойства, а строки представляют собой периоды, в определенной мере подобные друг другу.,
Таблица Д.И. Менделеева состоит из 8 групп. Физический смысл номера группы заключается в том, что номер группы соответствует валентности входящих в него элементов.
Если элемент обладает переменной валентностью, то номер группы соответствует наивысшей валентности данного элемента.
Каждая группа делится на главную (А) и побочную (В) подгруппу.
Главная подгруппа содержит элементы и малых, и больших периодов (S и p - элементы). Побочные подгруппы включают в себя d и f - элементы, т.е. элементы только больших периодов.
Элементы, составляющие одну подгруппу, имеют близкие химические свойства, т.к. имеют сходные валентные окончания. Химические свойства элементов обуславливаются их валентными окончаниями.
Валентное окончание элемента зависит от его положения в таблице Менделеева.
Все элементы одной подгруппы имеют одинаковую конфигурацию валентных окончаний, но разные квантовые числа.
С увеличением главного квантового числа у элементов данной подгруппы усиливаются металлические свойства, что обусловлено, в первую очередь, увеличением промежуточных электронных слоёв между ядром атома и его валентным слоем.
Элементы, составляющие одну группу, но разные подгруппы, имеют различные валентные окончания и различные химические свойства.
Энергия ионизации
Энергия (потенциал) ионизации атома Ei - минимальная энергия, необходимая для удаления электрона из атома на бесконечность в соответствии с уравнением
Х = Х+ + е−
Ее значения известны для атомов всех элементов Периодической системы. Например, энергия ионизации атома водорода соответствует переходу электрона с 1s-подуровня энергии (−1312,1 кДж/моль) на подуровень с нулевой энергией и равна +1312,1 кДж/моль.
В изменении первых потенциалов ионизации, соответствующих удалению одного электрона, атомов явно выражена периодичность при увеличении порядкового номера атома:
При движении слева направо по периоду энергия ионизации, вообще говоря, постепенно увеличивается, при увеличении порядкового номера в пределах группы - уменьшается. Минимальные первые потенциалы ионизации имеют щелочные металлы, максимальные - благородные газы.
Сродство к электрону
Сродство атома к электрону Ae - способность атомов присоединять добавочный электрон и превращаться в отрицательный ион. Мерой сродства к электрону служит энергия, выделяющая или поглощающаяся при этом. Сродство к электрону равно энергии ионизации отрицательного иона Х−:
Х− = Х + е−
Наибольшим сродством к электрону обладают атомы галогенов. Например, для атома фтора присоединение электрона сопровождается выделением 327,9 кДж/моль энергии. Для ряда элементов сродство к электрону близко к нулю или отрицательно, что значит отсутствие устойчивого аниона для данного элемента.
Обычно сродство к электрону для атомов различных элементов уменьшается параллельно с ростом энергии их ионизации.
Электроотрицательность
Электротрицательность характеризует способность атома химического элемента смещать в свою сторону электронное облако при образовании химической связи (в сторону элемента с более высокой электроотрицательностью). Американский физик Малликен предложил определять электроотрицательность как среднеарифметическую величину между потенциалом ионизации и сродством к электрону:
χ = 1/2 (Ei + Ae)
Трудность применения такого способа состоит в том, что значения сродства к электрону известны не для всех элементов.
Л. Полинг рекомендовал другой способ определения электроотрицательности. Он принял электроотрицательность фтора равной 4 (наибольшее значение), для цезия χ принимает наименьшее значение.
Электроотрицательность в количественном отношении представляет собой приближенную величину, поскольку она зависит от того, в состав какого конкретного соединения входит данный атом.
7) Классы неорганических соединений. Оксиды. Классификация оксидов и их химические свойства.(привести примеры реакций.)
Основные классы неорганических соединений и типы химических реакций
Основные классы неорганических соединений: оксиды, кислоты, основания и соли. Номенклатура неорганических соединений
Оксиды - это соединения элементов с кислородом. По химическим свойствам они подразделяются на солеобразующие и несолеобразующие. Солеобразующие оксиды в свою очередь подразделяются на основные, кислотные и амфотерные. Основным оксидам отвечают основания, кислотным - кислоты. Амфотерным оксидам отвечают гидраты, проявляющие и кислотные, и основные свойства.
Примерами основных оксидов могут служить оксид кальция СаО и оксид магния MgO. Оксид кальция взаимодействует с водой, образуя гидро-ксид кальция Са(ОН)2:
СаО + Н2О = Са(ОН)2.
Оксид магния малорастворим в воде; однако ему соответствует основание - гидроксид магния Mg(OH)2, который можно получить из оксида магния косвенным путем.
Примерами кислотных оксидов могут служить триоксид серы SO3и диоксид кремния SiO2. Первый из них взаимодействует с водой, образуя серную кислоту H2SO4:
SO3 + Н2О = H2SO4.
Диоксид кремния с водой не взаимодействует, но ему соответствует кремниевая кислота H2SiО3, которую можно получить из SiO2косвенным путем.
Кислотные оксиды можно получить из кислот, отнимая от них воду. Поэтому их называют также ангидридами кислот или просто ангидридами.
К несолеобразующим оксидам относится, например, оксид азота (I) N2О. Нет такой кислоты или основания, которые отвечали бы этому оксиду.
Существуют различные номенклатуры оксидов. До сих пор в промышленности могут использоваться устаревшие термины русской номенклатуры.
Согласно международной номенклатуре (которой пользуются в настоящее время и отечественные химики) все соединения элементов с кислородом (за исключением пероксидов) называются оксидами. При этом для элементов переменной валентности в скобках римскими цифрами указывается валентность, которую элемент проявляет в данном оксиде. Так, СаО называется оксид кальция, а Сu2О и СuО - оксид меди (I), оксид меди (II). По отечественной номенклатуре оксиды состава ЭО2или ЭО3называют также, соответственно, диоксидами и триоксидами.
Согласно устаревшей отечественной номенклатуре, если элемент образует только один оксид, то последний назывался окисью. Так, СаО назывался окисью кальция. Если существует два или несколько оксидов данного элемента, то их названия образовывались в соответствии с числом атомов кислорода, приходящихся на один атом элемента, например: Э2О - полуокись, ЭО - одноокись, Э2О3- полутораокись, ЭО2- двуокись, Э2О5- полупятиокись, ЭО3- трехокись (символом Э здесь обозначен атом соответствующего элемента). Так, FeO -одноокись железа, Fe2O3- полутораокись железа, Сu2О - полуокись меди, СuО - одноокись меди. Иногда оксиды, в которых элемент проявляет низшую валентность, назывались закисями (Сu2О - закись меди, N2O - закись азота), а кислотные оксиды - ангидридами соответствующих кислот (N2O5-азотный ангидрид, Мn2O7- марганцовый ангидрид).
Существуют вещества - соединения элементов с кислородом - лишь формально принадлежащие к классу оксидов. К таким веществам относятся, в частности, пероксиды (перекиси) металлов, например, пероксид (перекись) бария ВаО2. По своей природе подобные вещества представляют собой соли очень слабой кислоты - пероксида (перекиси) водорода Н2О2.
Основания состоят из металла и одновалентных гидроксогрупп ОН, число которых равно валентности металла. Примерами оснований могут служить гидроксид натрия NaOH, гидроксид меди Сu(ОН)2.
8) Классы неорганических соединений. Кислоты и их классификация. Получение и химические свойства кислот. (привести примеры реакций.)
Кислоты состоят из водорода, способного замещаться металлом, и кислотного остатка, причем число атомов водорода равно валентности кислотного остатка. Примерами кислот могут служить соляная (хлористоводородная) НСl, серная H2SO4, азотная HNO3, уксусная СН3СООН.
Важнейшее химическое свойство кислот - их способность образовывать соли с основаниями. Например, при взаимодействии кислот c гидроксидом натрия получаются натриевые соли этих кислот:
2NaOH + H2SO4 = Na2SO4 + 2H2O; NaOH + HNO3 = NaNO3 + H2O.
Кислоты классифицируются по их силе, по основности и по наличию кислорода в составе кислоты. По силе кислоты делятся на сильные и слабые. Важнейшие сильные кислоты - это азотная, серная и соляная.
Основностью кислоты называется число атомов водорода в молекуле кислоты, способных замещаться на металл с образованием соли. Такие кислоты, как соляная и уксусная, могут служить примерами одноосновных кислот, серная кислота - двухосновна, ортофосфорная кислота Н3РО4 - трехосновна.
По наличию кислорода в своем составе кислоты делятся на кислородсодержащие и бескислородные. Азотная и серная кислоты - кислородсодержащие кислоты, соляная кислота и сероводород - бескислородные.
Названия кислот производят от того элемента, от которого образована кислота. При этом названия бескислородных кислот имеют окончание водородная: НСl - хлороводородная (соляная кислота), H2S - сероводородная, HCN - циановодородная (синильная кислота). Названия кислородсодержащих кислот также образуются от названия соответствующего элемента с добавлением слова кислота: HNO3- азотная, Н2CrO4- хромовая. Если элемент образует несколько кислот, то различие между ними отражается в окончаниях их названий. Название кислоты, в которой элемент проявляет высшую валентность, оканчивается на ная или овая; если же валентность элемента ниже максимальной, то название кислоты оканчивается на истая или овистая. Например, НNO3- азотная кислота, HNO2- азотистая, Н3AsO4- мышьяковая, H3AsO3- мышьяковистая. Кроме того, одному и тому же оксиду могут отвечать несколько кислот, различающихся между собой числом молекул воды. При этом наиболее богатая водой форма имеет приставку орто, а наименее богатая - мета. Так, кислота Н3РО4, в которой на одну молекулу фосфорного ангидрида Р2О5приходится три молекулы воды, называется ортофосфорная, а кислота НРО3- метафосфорная, так как в ней на одну молекулу Р2О5приходится одна молекула воды. Указанная номенклатура кислот не строга. Наряду с приведенными окончаниями и приставками употребляются и другие. Кроме того, ряд кислот имеют исторически сложившиеся названия.
9) Классы неорганических соединений. Основания и амфотерные гидрооксиды. Способы получения и их химические свойства. (привести примеры реакций.)
Важнейшее химическое свойство оснований - способность образовывать с кислотами соли. Например, при взаимодействии перечисленных оснований с соляной кислотой получаются хлористые соли соответствующих металлов - хлориды натрия или меди:
NaOH + НС1 = NaCl + Н2О; Cu(OH)2 + 2НС1 = CuCl2+ 2Н2О.
Основания классифицируют по их растворимости в воде и по их силе. По растворимости основания делятся на растворимые, или щелочи, и на нерастворимые. Важнейшие щелочи - это гидроксиды натрия, калия и кальция. По силе основания делятся на сильные и слабые. К сильным относятся все щелочи, кроме гидроксида аммония. Согласно международной номенклатуре соединения, содержащие в своем составе гидроксогруппы, называют гидроксидами. В случае металлов переменной валентности в скобках указывают валентность металла в данном соединении. Так, Са(ОН)2 - гидроксид кальция, Fe(OH)2- гидроксид железа (II), Fe(OH)3 - гидроксид железа (III).
В устаревшей русской номенклатуре названия оснований обычно образовывались, прибавлением к названию соответствующего оксида приставку гидро- или слово гидрат. Так, Са(ОН)2- гидроокись кальция, Fe(OH)2- гидрат закиси железа, Fe(OH)3 - гидроокись или гидрат окиси железа.
10) Классы неорганических соединений. Соли. Классификация солей. Способы получений средних,кислых, основных солей, их названия. (привести примеры реакций.)
Продукты замещения водорода в кислоте на металл или гидроксогрупп в основании на кислотный остаток представляют собою соли. При полном замещении получаются средние (нормальные) соли, при неполном - или кислые, или основные. Кислая соль получается при неполном замещении водорода кислоты на металл. Основная соль получается при неполном замещении гидроксогрупп основания на кислотный остаток. Ясно, что кислая соль может быть образована только кислотой, основность которой равна двум или больше, а основная соль - металлом, валентность которого равна двум или больше.
Примеры образования солей:
Са(ОН)2+ H2SO4= СаSO4+ 2Н2О,
СаSO4- нормальная соль - сульфат кальция; КОН + H2SO4= KHSO4+ Н2О,
KHSO4 - кислая соль - гидросульфат калия;
Mg(OH)2+ HC1 = MgOHCl + Н2О,
MgOHCl - основная соль - хлорид гидроксомагния.
Соли, образованные двумя металлами и одной кислотой, называются двойными солями; соли, образованные одним металлом и двумя кислотами - смешанными солями. Примером двойной соли могут служить алюмокалиевые квасцы, или сульфат калия-алюминия, KAI(SO4)2. Смешанной солью является CaClOCl или (CaOCl2) - кальциевая соль соляной (HCl) и хлорноватистой (HClО) кислот.
Одна и та же соль может называться по-разному. Например, KNO3называют калиевой селитрой, азотнокалиевой солью, азотнокислым калием, нитратом калия. Сейчас большинство химиков пользуются для солей международной (латинской) номенклатурой. В этой номенклатуре название соли отражает название металла и латинское название кислотного остатка. Латинское название кислоты и кислотного остатка происходит обычно от латинского названия элемента, образующего кислоту. При этом название соли бескислородной кислоты имеет окончание ид, кислородсодержащей кислоты - am в случае максимальной валентности кислотообразующего элемента и ит в случае более низкой его валентности. Так, соли соляной кислоты называются хлориды, сероводородной - сульфиды, серной - сульфаты и сернистой - сульфиты.
Для солей, образованных металлами с переменной валентностью, валентность металла указывают в скобках, как в оксидах или основаниях: так, FeSO4- сульфат железа (II), Fe2(SO4) - сульфат железа (III). Название кислой соли имеет приставку гидро, указывающую на наличие незамещенных атомов водорода; если таких незамещенных атомов два или больше, то их число обозначается греческими числительными (ди-, три- и т.д.). Так, Na2HPO4 называется гидрофосфатом натрия, a NaH2PO4- дигидрофосфатом натрия. Аналогично основная соль характеризуется приставкой гидроксо, указывающей на наличие незамещенных гидроксильных групп. Например, AlOHCl2называется хлоридом гидроксоалюминия, Аl(ОН)2С1 - хлоридом дигидроксоалюминия.
11) Химическая система. Гомогенные и гетерогенные системы. Термодинамические параметры и термодинамические функции состояния. Первый закон термодинамики. Внутренняя энергий и энтальпия.
Гомоге́нная систе́ма(от греч. ὁμός — равный, одинаковый; γένω — рождать) — однородная система, химический состав и физические свойства которой во всех частях одинаковы или меняются непрерывно, без скачков (между частями системы нет поверхностей раздела). В гомогенной системе из двух и более химических компонентов каждый компонент распределен в массе другого в виде молекул, атомов, ионов. Составные части гомогенной системы нельзя отделить друг от друга механическим путем.
В гомогенных смесях составные части нельзя обнаружить ни визуально, ни с помощью оптических приборов, поскольку вещества находятся в раздробленном состоянии на микроуровне. Гомогенными смесями являются смеси любых газов и истинные растворы, а также смеси некоторых жидкостей и твердых веществ, например сплавы.
Гетероге́нная систе́ма(от греч. ἕτερος — разный; γένω — рождать) — неоднородная система, состоящая из однородных частей (фаз), разделенных поверхностью раздела. Однородные части (фазы) могут отличаться друг от друга по составу и свойствам. Число веществ (компонентов), термодинамических фаз и степеней свободы связаны правилом фаз. Примерами гетерогенных систем могут служить: жидкость — насыщенный пар; насыщенный раствор с осадком; многие сплавы. Твердый катализатор в токе газа или жидкости тоже гетерогенная система (гетерогенный катализ).
Первый закон термодинамики(закон сохранения энергии для тепловых процессов) определяет количественное соотношение между изменением внутренней энергии системы дельта U, количеством теплоты Q, подведенным к ней, и суммарной работой внешних сил A, действующих на систему.
![]()
Первый закон термодинамики - Изменение внутренней энергии системы при ее переходе из одного состояния в другое равно сумме количества теплоты, подведенного к системе извне, и работы внешних сил, действующих на нее:
![]()
Первый закон термодинамики - количество теплоты, подведенное к системе, идет на изменение ее внутренней энергии и на совершение системой работы над внешними телами:
Термодинамическая функция состояния— в термодинамике некая функция, зависящая от нескольких независимых параметров, которые однозначно определяют состояние термодинамической системы. Значение термодинамической функции состояния зависит только от состояния термодинамической системы и не зависит от того, как система пришла в это состояние. Частным случаем функций состояний являются термодинамические потенциалы.
Внутренняя энергия тела— это сумма энергий молекулярных взаимодействий и тепловых движений молекулы. Внутренняя энергия является однозначной функцией состояния системы. Это означает, что всякий раз, когда система оказывается в данном состоянии, её внутренняя энергия принимает присущее этому состоянию значение, независимо от предыстории системы. Следовательно, изменение внутренней энергии при переходе из одного состояния в другое будет всегда равно разности между ее значениями в конечном и начальном состояниях, независимо от пути, по которому совершался переход.
Энтальпия, также тепловая функция и теплосодержание — термодинамический потенциал, характеризующий состояние системы в термодинамическом равновесии при выборе в качестве независимых переменных давления, энтропии и числа частиц.
Проще говоря, энтальпия - это та энергия, которая доступна для преобразования в теплоту при определенных температуре и давлении.
Энтальпией образования химических соединенияназывают энтальпию реакции образования данного соединения из простых веществ. В качестве простых веществ выбирают химические элементы в их естественном фазовом и химическом состоянии при данной температуре.
Так, при 298 К для хлора простым веществом служит газообразный хлор, состоящий из молекул С12, а для калия - металлический калий. Энтальпия образования твердого КСl при 298 К - это энтальпия реакции: К(тв.) +1/2Сl2= КСl(тв.). Энтальпия образования КСl (тв.) при 500 К соответствует реакции: К(жидкий) +1/2Cl2= КСl(тв.), так как естественным фазовым состоянием (простым веществом) для калия становится уже не кристалл, а жидкость (т-ра плавления К 336,66 К).
12) Термохимия. Термохимические уравнения и расчёты. Закон Гесса и следствие из него.
Термохимия раздел химической термодинамики, в задачу которой входит определение и изучение тепловых эффектов реакций, а также установление их взваимосвязей с различными физико-химическими параметрами. Ещё одной из задач термохимии является измерение теплоёмкостей веществ и установление их теплот фазовых переходов.
Термохимические уравнения. Теплота, высвобождаемая или поглощаемая конкретной химической реакцией, пропорциональна степени превращения реагентов, определяемой по количеству любого из расходуемых либо образующихся продуктов. Изменение внутренней энергии или энтальпии реагирующей системы определяют по химическому уравнению реакции. Например, сгорание смеси газообразных метана и кислорода описывается термохимическим уравнением:
CH4(г) + 2O2 (г)=CO2 (г) + H2O (ж);
^H° = -212,768 ккал.
Здесь буквы в скобках обозначают агрегатные состояния веществ (газ или жидкость). Символом ^H° обозначается изменение энтальпии в химическом превращении при стандартных давлении 1 атм и температуре 298 K (25° С) Химическая формула каждого вещества в таком уравнении обозначает вполне определенное количество вещества, а именно его молекулярную массу, выраженную в граммах. Молекулярная масса получается сложением атомных масс всех элементов, входящих в формулу, с коэффициентами, равными числу атомов данного элемента в молекуле. Молекулярная масса метана равна 16,042, и, согласно предыдущему уравнению, при сгорании 16,042 г (1 моля) метана получаются продукты, энтальпия которых на 212,798 ккал меньше энтальпии реагентов. В соответствии с уравнением (5) такое количество теплоты высвобождается, когда 1 моль метана сгорает в кислороде при постоянном давлении 1 атм. Соответствующее уменьшение внутренней энергии системы в ходе реакции составляет 211,615 ккал. Разница между DH° и DU° равна –1,183 ккал и представляет работу p(V2 – V1), совершаемую, когда 3 моля газообразных реагентов сжимаются при давлении 1 атм до 1 моля газообразного диоксида углерода и 2 молей жидкой воды.
Закон Гесса — основной закон термохимии, который формулируется следующим образом:
Тепловой эффект химической реакции зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Иными словами, количество теплоты, выделяющееся или поглощающееся при каком-либо процессе, всегда одно и то же, независимо от того, протекает ли данное химическое превращение в одну или в несколько стадий (при условии, что температура, давление и агрегатные состояния веществ одинаковы). Закон открыт русским химиком Г.И. Гессом в 1840 г.; он является частным случаем первого начала термодинамики применительно к химическим реакциям. Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты самых разнообразных химических процессов; для этого обычно используют ряд следствий из него.
Следствия из закона Гесса:
*Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье — Лапласа).
*Тепловой эффект химической реакции равен разности сумм теплот образования (?Hf) продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты (?):
*Тепловой эффект химической реакции равен разности сумм теплот сгорания (?Hc) исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты (?):
Таким образом, пользуясь табличными значениями теплот образования или сгорания веществ, можно рассчитать теплоту реакции, не прибегая к эксперименту.
13) Второй закон термодинамики. Понятие об энтропии. Изменение энтропии при химических и фазовых процессах. Изобарно-изотермический потенциал(энергия Гиббса) как критерий возможности самопроизвольного протекания процесса.
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
Формулировки
Существуют несколько эквивалентных формулировок второго начала термодинамики:
Постулат Клаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему» (такой процесс называется процессом Клаузиуса).
Постулат Томсона: «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
Эквивалентность этих формулировок легко показать. В самом деле, допустим, что постулат Клаузиуса неверен, то есть существует процесс, единственным результатом которого была бы передача тепла от более холодного тела к более горячему. Тогда возьмем два тела с различной температурой (нагреватель и холодильник) и проведем несколько циклов тепловой машины забрав тепло Q1 у нагревателя, отдав Q2 холодильнику и совершив при этом работу A = Q1 ? Q2. После этого воспользуемся процессом Клаузиуса и вернем тепло Q2 от холодильника нагревателю. В результате получается, что мы совершили работу только за счет отъёма теплоты от нагревателя, то есть постулат Томсона тоже неверен.
С другой стороны, предположим, что неверен постулат Томсона. Тогда можно отнять часть тепла у более холодного тела и превратить в механическую работу. Эту работу можно превратить в тепло, например, с помощью трения, нагрев более горячее тело. Значит, из неверности постулата Томсона следует неверность постулата Клаузиуса.
Таким образом, постулаты Клаузиуса и Томсона эквивалентны.
Другая формулировка второго начала термодинамики основывается на понятии энтропии:
«Энтропия изолированной системы не может уменьшаться» (закон неубывания энтропии).
Такая формулировка основывается на представлении об энтропии как о функции состояния системы, что также должно быть постулировано.
В состоянии с максимальной энтропией макроскопические необратимые процессы (а процесс передачи тепла всегда является необратимым из-за постулата Клаузиуса) невозможны.
Термодинамическая энтропия S, часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы; её существование постулируется вторым началом термодинамики.
Термодинамическое определение энтропии:
Понятие энтропии было впервые введено в 1865 году Рудольфом Клаузиусом. Он определил изменение энтропии термодинамической системы при обратимом процессе как отношение изменения общего количества тепла ?Q к величине абсолютной температуры T:
^S = ^Q/T
Рудольф Клаузиус дал величине S имя «энтропия», происходящее от греческого слова ??o??, «изменение» (изменение, превращение, преобразование). Данное равенство относится к изменению энтропии, не определяя полностью саму энтропию.
Эта формула применима только для изотермического процесса (происходящего при постоянной температуре). Её обобщение на случай произвольного квазистатического процесса выглядит так:
dS = ?Q/T
где dS - приращение (дифференциал) энтропии, а ?Q - бесконечно малое приращение количества теплоты.
Необходимо обратить внимание на то, что рассматриваемое термодинамическое определение применимо только к квазистатическим процессам (состоящим из непрерывно следующих друг за другом состояний равновесия).
Поскольку энтропия является функцией состояния, в левой части равенства стоит её полный дифференциал. Напротив, количество теплоты является функцией процесса, в котором эта теплота была передана, поэтому ?Q считать полным дифференциалом нельзя.
Энтропия, таким образом, согласно вышеописанному, определена вплоть до произвольной аддитивной постоянной. Третье начало термодинамики позволяет определить её точнее: предел величины энтропии равновесной системы при стремлении температуры к абсолютному нулю полагают равным нулю.
Энтропия (S)– термодинамическая функция состояния, которая служит мерой беспорядка (неупорядоченности) системы. Возможность протекания эндотермических процессов обусловлена изменением энтропии, ибо в изолированных системах энтропия самопроизвольно протекающего процесса увеличивается ΔS > 0 (второй закон термодинамики)
Энергия Гиббса(изобарно-изотермический потенциал). Во многих случаях самопроизвольные процессы (процессы, происходящие без подвода энергии от внешнего источника) в природе протекают при наличии разности потенциалов, например, разность электрических потенциалов, обусловливает перенос заряда, а разность гравитационных потенциалов – падение тела. Эти процессы заканчиваются при достижении минимума потенциала. Движущей силой химических процессов, протекающих при постоянных давлении и температуре является изобарно-изотермический потенциал, называемый в настоящее время энергией Гиббса и обозначаемый G. Изменение энергии Гиббса в химическом процессе определяется соотношением
ΔG = ΔH –TΔS,
где ΔG – изменение энергии Гиббса химического процесса; ΔH – изменение энтальпии химического процесса; ΔS – изменение энтропии химического процесса; Т – температура в Кельвинах.
Уравнение (2.16) может быть представлено в следующем виде: ΔH = ΔG + TΔS. (2.17)
Смысл уравнения (2.17) в том, что часть теплового эффекта реакции расходуется на совершение работы (ΔG), а часть рассеивается в окружающую среду (TΔS).
Энергия Гиббса является критерием принципиальной возможности самопроизвольного протекания реакции. Если в ходе реакции энергия Гиббса уменьшается, то процесс может протекать в данных условиях самопроизвольно ΔG < 0. Процесс в данных условиях неосуществим, если ΔG > 0. Реакция является обратимой, т.е. может протекать и в прямом и в обратном направлении, если ΔG = 0 (термодинамическое условие химического равновесия).
Эти соотношения применимы также к фазовым равновесиям, т.е. случаям, когда в равновесии находятся две фазы (агрегатных состояния) одного и того же вещества, например, лед и жидкая вода.
Энтальпийный и энтропийный факторы. Процессы могут протекать самопроизвольно (ΔG<0), если они сопровождаются уменьшением энтальпии (ΔH<0) и увеличением энтропии системы (ΔS>0). Если же энтальпия системы увеличивается (ΔH>0), а энтропия уменьшается (ΔS<0), то такой процесс протекать не может (ΔG>0). При иных знаках ΔS и ΔН принципиальная возможность протекания процесса определяется соотношением энтальпийного (ΔH) и энтропийного (ТΔS) факторов.
Если ΔН>0 и ΔS>0, т.е. энтальпийная составляющая противодействует, а энтропийная благоприятствует протеканию процесса, то реакция может протекать самопроизвольно за счет энтропийной составляющей, при условии, что |ΔH|<|TΔS|.
Если, энтальпийная составляющая благоприятствует, а энтропийная противодействует протеканию процесса, то реакция может протекать самопроизвольно за счет энтальпийной составляющей, при условии, что |ΔH|>|TΔS|.
Влияние температуры на направление реакции. Изменение знака энергии Гиббса произойдет при
 ( (2.21)
( (2.21)
Очевидно, что смена знака энергии Гиббса с изменением температуры возможна только в двух случаях: 1) ΔН>0 и ΔS>0 и 2) ΔН<0 и ΔS<0.
Стандартная энергия Гиббса образования
-
 это изменение энергии Гиббса реакции
образования 1 моль соединения из простых
веществ, устойчивых при стандартных
условиях. Энергия Гиббса образования
простых веществ принимается равной
нулю. Стандартные энергии Гиббса
образования веществ можно найти в
соответствующих справочниках.
это изменение энергии Гиббса реакции
образования 1 моль соединения из простых
веществ, устойчивых при стандартных
условиях. Энергия Гиббса образования
простых веществ принимается равной
нулю. Стандартные энергии Гиббса
образования веществ можно найти в
соответствующих справочниках.
Энергия Гиббса химической реакции. Энергия Гиббса является функцией состояния, т.е. ее изменение в процессе не зависит от пути его протекания, а определяется исходным и конечным состоянием системы. Следовательно, энергию Гиббса химической реакции (2.10) можно рассчитать по формуле

Если условия отличаются от стандартных, то для нахождения ΔrG может быть использовано уравнение изотермы Вант-Гоффа, которое для реакции (2.11) между газами записывается как

а между растворенными веществами -
где
 _ относительные парциальные давления
соответствующих веществ; сА, сВ,
сD, cE_ концентрации
соответствующих растворенных веществ,
а, b, c, d – соответствующие стехиометрические
коэффициенты.
_ относительные парциальные давления
соответствующих веществ; сА, сВ,
сD, cE_ концентрации
соответствующих растворенных веществ,
а, b, c, d – соответствующие стехиометрические
коэффициенты.
14) Химическое равновесие. Константа равновесия. Основное уравнение термодинамики.
Химическое равновесие — состояние химической системы, при котором возможны реакции, идущие с равными скоростями в противоположных направлениях. При химическом равновесии концентрации реагентов, температура и другие параметры системы не изменяются со временем.
Все химические реакции, в принципе, обратимы. Это означает, что в реакционной смеси протекает как взаимодействие реагентов, так и взаимодействие продуктов. В этом смысле различие между реагентами и продуктами условное. Направление протекания химической реакции определяется условиями ее проведения (температурой, давлением, концентрацией веществ). Многие реакции имеют одно преимущественное направление и для проведения таких реакций в противоположном направлении требуются экстремальные условия. В подобных реакциях происходит почти полное превращение реагентов в продукты.
В состоянии равновесия скорости прямой и обратной реакции становятся равными.
СМЕЩЕНИЕ ХИМИЧЕСКОГО РАВНОВЕСИЯ:
Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации.
КОНСТАНТА ХИМИЧЕСКОГО РАВНОВЕСИЯ - величина, выражающая соотношение между концентрациями (парциальными давлениями, летучестями, активностями) компонентов системы в состоянии химического равновесия. Численные значения химического равновесия константы позволяют рассчитывать выход продуктов реакции в данных условиях по начальным концентрациям реагирующих веществ.
Таким образом, константа равновесия состоит из двух множителей, один из которых содержит энтальпию реакции (или, при определенных условиях, внутреннюю энергию реакции), а другой – энтропию.
Важное следствие общей термодинамической теории состоит в том, что можно вывести уравнение, связывающее зависимость DG° (и следовательно, К) от температуры со значением DH°. Поэтому можно точно установить направление и равновесное состояние химической реакции при одной температуре из известных данных по химическому равновесию при другой температуре. В частности, изменение DG° с температурой пропорционально величине DH° и имеет противоположный знак. Так, все эндотермические реакции (DH° положительна) ускоряются (большая отрицательная величина DG° и большее значение К) с ростом температуры. Соответственно все экзотермические реакции замедляются с ростом температуры.
15) Химическое равновесие. Принцип Ла-Шателье. Сдвиг химического равновесия на примере реакции синтеза аммиака.
Химическое равновесие — состояние химической системы, при котором возможны реакции, идущие с равными скоростями в противоположных направлениях. При химическом равновесии концентрации реагентов, температура и другие параметры системы не изменяются со временем.
КОНСТАНТА ХИМИЧЕСКОГО РАВНОВЕСИЯ - величина, выражающая соотношение между концентрациями (парциальными давлениями, летучестями, активностями) компонентов системы в состоянии химического равновесия. Численные значения химического равновесия константы позволяют рассчитывать выход продуктов реакции в данных условиях по начальным концентрациям реагирующих веществ.
Ле Шателье — Брауна принцип (принцип смещения равновесия), устанавливает, что внешнее воздействие, выводящее систему из состояния термодинамического равновесия, вызывает в системе процессы, стремящиеся ослабить эффект воздействия. Так, при нагревании равновесной системы в ней происходят изменения (например, химические реакции), идущие с поглощением теплоты, а при охлаждении — изменения, протекающие с выделением теплоты. При увеличении давления смещение равновесия связано с уменьшением общего объёма системы, а уменьшению давления сопутствуют физические или химические процессы, приводящие к увеличению объема.
Влияние температуры. В каждой обратимой реакции одно из направлений отвечает экзотермическому процессу, а другое - эндотермическому.
N2 + 3H2 = 2NH3 + Q
Прямая реакция - экзотермическая, а обратная реакция - эндотермическая.
Влияние изменения температуры на положение химического равновесия подчиняется следующим правилам: При повышении температуры химическое равновесие смещается в направлении эндотермической реакции, при понижении температуры - в направлении экзотермической реакции.
Влияние давления Во всех реакциях с участием газообразных веществ, сопровождающихся изменением объема за счет изменения количества вещества при переходе от исходных веществ к продуктам, на положение равновесия влияет давление в системе.
Влияние давления на положение равновесия подчиняется следующим правилам: При повышении давления равновесие сдвигается в направлении образования веществ (исходных или продуктов) с меньшим объемом; при понижении давления равновесие сдвигается в направлении образования веществ с большим объемом:
N2 + 3H2 = 2NH3
Таким образом, при переходе от исходных веществ к продуктам объем газов уменьшился вдвое. Значит, при повышении давления равновесие смещается в сторону образования NH3.
Влияние концентрации Влияние концентрации на состояние равновесия подчиняется следующим правилам:
При повышении концентрации одного из исходных веществ равновесие сдвигается в направлении образования продуктов реакции;
При повышении концентрации одного из продуктов реакции равновесие сдвигается в направлении образования исходных веществ.
На состояние химического равновесия оказывают влияние концентрация реагирующих веществ, температура, а для газообразных веществ - и давление. При изменении одного из этих параметров равновесие нарушается и концентрация всех реагирующих веществ изменяется до тех пор, пока не установится новое равновесие, но уже при иных значениях равновесных концентраций. Подобный переход реакционной системы от одного состояния равновесия к другому называется смещением (или сдвигом) химического равновесия. Если при изменении условий увеличивается концентрация конечных веществ, то говорят о смещении равновесия в сторону продуктов реакции. Если же увеличивается концентрация исходных веществ, то равновесие смещается в сторону их образования.
16) Химическая связь. Виды химической связи. Межмолекулярное взаимодействие. Ковалентная связь. Метод валентных связей. Направление ковалентной связи. Механизмы образования связей.
Химическая связь - это взаимодействие, которое связывает отдельные атомы в молекулы, ионы, радикалы, кристаллы.
Основным условием образования химической связи является понижением полной энергии многоатомной системы по сравнению с энергией изолированных атомов, т.е. ЕАВЕА+ЕВ в случае образования вещества АВ из А и В. Более точно химическую связь можно определить как взаимодействие атомов, обусловленное перекрыванием их электронных облаков, и сопровождается уменьшением полной энергии системы.
Основными параметрами химической связи является её длина, прочность и валентные углы, характеризующие строение веществ, которые образованы из отдельных атомов.
Длина связи - это межъядерное расстояние между химическими связанными атомами.
Угол между воображаемыми прямыми, проходящими через ядра химически связанных атомов, называется валентным углом. Энергия связи - энергия, необходимая для разрыва такой связи.
Химическая связь
I. Ионная
II. Ковалентная.
III. Координационная.
IV. Металлическая.
V. Водородная.
VI. Межмолекулярная.
Ковалентная связь (атомная связь, гомеополярная связь) — химическая связь, образованная перекрытием (обобществлением) пары валентных электронных облаков. Обеспечивающие связь электронные облака (электроны) называются общей электронной парой.
Характерные свойства ковалентной связи — направленность, насыщаемость, полярность, поляризуемость — определяют химические и физические свойства соединений.
Направленность связи обусловлена молекулярным строением вещества и геометрической формы их молекулы. Углы между двумя связями называют валентными.
Насыщаемость — способность атомов образовывать ограниченное число ковалентных связей. Количество связей, образуемых атомом, ограничено числом его внешних атомных орбиталей.
Полярность связи обусловлена неравномерным распределением электронной плотности вследствие различий в электроотрицательностях атомов. По этому признаку ковалентные связи подразделяются на неполярные и полярные.
Поляризуемость связи выражается в смещении электронов связи под влиянием внешнего электрического поля, в том числе и другой реагирующей частицы. Поляризуемость определяется подвижностью электронов. Полярность и поляризуемость ковалентных связей определяет реакционную способность молекул по отношению к полярным реагентам.
ВАЛЕНТНЫХ СВЯЗЕЙ МЕТОД (метод валентных схем), метод приближенного решения электронного ур-ния Шрёдингера для многоэлектронных молекулярных систем. Основан на представлениях о двухцентровых хим. связях между атомами в молекуле, образуемых двумя электронами. Эти представления являются обобщением на многоатомные молекулы приближения Гайтлера - Лондона, позволившего впервые с помощью квантовомех. методов объяснить хим. связь в молекуле Н2.
Осн.
физ. идея В. с. м. состоит в том, что
волновая ф-ция молекулы выражается
через волновые ф-ции составляющих ее
атомов. Образование хим. связи
рассматривается как результат спаривания
спинов своб. электронов атомов. Тем
самым В. с. м. дает обоснование одному
из осн. положений теории валентности:
валентность нейтрального атома равна
числу своб. электронов в его валентной
оболочке. Каждому валентному штриху,
соединяющему атомы А и В в структурной
ф-ле молекулы, отвечает двухэлектронная
ф-ция валентной связи ХАВ(1,2),
к-рая представляется в виде произведения
двух волновых ф-ций: пространственной
Ф(1,2), симметричной относительно
перестановки координат электронов, и
спиновой (1,2),
антисимметричной относительно такой
перестановки и описывающей систему
двух электронов с противоположными
спинами; цифры 1 и 2 в этих обозначениях
указывают пространств. координаты или
спиновые переменные первого и второго
электронов либо те и другие одновременно.
(1,2),
антисимметричной относительно такой
перестановки и описывающей систему
двух электронов с противоположными
спинами; цифры 1 и 2 в этих обозначениях
указывают пространств. координаты или
спиновые переменные первого и второго
электронов либо те и другие одновременно.

Для
простейшей молекулы Н2
ф-цию Ф(1,2) строят из 1s-орбиталей атомов
Н, обозначаемых для разных ядер как и
и ,
а ф-цию
,
а ф-цию (1,2)
- из одноэлектронных спиновых
ф-ций
(1,2)
- из одноэлектронных спиновых
ф-ций и
и (спин-функций),
описывающих состояния электронов с
противоположно направленными спинами:
(спин-функций),
описывающих состояния электронов с
противоположно направленными спинами:

Основное:
1) Ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам.
2) Ковалентная связь тем прочнее, чем в большей степени перекрываются электронные облака.
Простая ковалентная связь образуется из двух неспаренных валентных электронов, на один
от каждого атома:
A· + ·В → А : В
В результате обобществления электроны образуют заполненный энергетический уровень. Связь образуется, если их суммарная энергия на этом уровне будет меньше, чем в первоначальном состоянии (а разница в энергии будет не чем иным, как энергией связи).
Заполнение электронами атомных (по краям) и молекулярных (в центре) орбиталей в молекуле H2. Вертикальная ось соответствует энергетическому уровню, электроны обозначены стрелками, отражающими их спины.
Согласно теории молекулярных орбиталей, перекрывание двух атомных орбиталей приводит в простейшем случае к образованию двух молекулярных орбиталей (МО): связывающей МО и антисвязывающей (разрыхляющей) МО. Обобществленные электроны располагаются на более низкой по энергии связывающей МО.
17) Метод валентных связей. Гибридизация атомных орбиталей. Типы гибридизации на примере BeH2, BH3, CH4, H2O.
Представления о механизме образования молекулы водорода были распространены на более сложные молекулы. Разработанная на этой основе теория химической связи получила название метода валентных связей (метод ВС). В основе метода ВС лежат следующие положения:
1) Ковалентная связь образуется двумя электронами с противоположно направленными спинами, причем эта электронная пара принадлежит двум атомам.
2) Ковалентная связь тем прочнее, чем в большей степени перекрываются электронные облака.
Комбинации двухэлектронных двухцентровых связей, отражающие электронную структуру молекулы, получили название валентных схем. Примеры построения валентных схем:


В валентных схемах наиболее наглядно воплощены представления Льюиса об образовании химической связи путем обобществления электронов с формированием электронной оболочки благородного газа: для водорода – из двух электронов (оболочка He), для азота – из восьми электронов (оболочка Ne).
ВАЛЕНТНЫХ СВЯЗЕЙ МЕТОД (метод валентных схем), метод приближенного решения электронного ур-ния Шрёдингера для многоэлектронных молекулярных систем. Основан на представлениях о двухцентровых хим. связях между атомами в молекуле, образуемых двумя электронами. Эти представления являются обобщением на многоатомные молекулы приближения Гайтлера - Лондона, позволившего впервые с помощью квантовомех. методов объяснить хим. связь в молекуле Н2.
Гибридизация атомных орбиталей, квантовохимический способ описания перестройки орбиталей атома в молекуле по сравнению со свободным атомом. Являясь формальным математическим приемом, гибридизация атомных орбиталей позволяет отразить нарушение сферической симметрии распределения электронной плотности атома при образовании химической связи.
Сущность
гибридизации атомных орбиталей состоит
в том, что электрон молекулы вблизи
выделенногибридная орбиталь атомногибридная
орбиталь ядра характеризуется не
отдельной атомной орбиталью, а линейной
комбинацией атомных орбиталей с различным
значениями азимутальногибридная
орбиталь и магнитногибридная орбиталь
квантовых чисел. Такая линейная комбинация
называется гибридной (гибридизированной)
орбиталью. Как правило, гибридизация
затрагивает лишь высшие и близкие по
энергии занятые атомные орбитали
свободногибридная орбиталь атома.
Например, для атомов элементов
второгибридная орбиталь периода
периодической системы типичная форма
гибридной орбитали
 -
линейная комбинация 2s-орбитали
-
линейная комбинация 2s-орбитали
 и
2р-орбиталей
и
2р-орбиталей

 ,
,

 ,
,
 с
численными коэффициентами.
с
численными коэффициентами.
Гибридные
орбитали обладают более низкой симметрией,
чем составляющие их атомные орбитали.
Так, распределение электронной плотности,
отвечающее указанной гибридной орбитали
 ,
смещено от атомного ядра в направлении
вектора п
с координатами
,
смещено от атомного ядра в направлении
вектора п
с координатами
 ;
вектор п
является
осью симметрии гибридной орбитали (рис.
1). При изменении ориентации в пространстве
осей координат коэффициенты линейной
комбинации могут изменяться, однако
остается постоянным отношение сумм
квадратов коэффициентов для данного
значения азимутального квантового
числа. Это отношение определяет тип
гибридной орбитали.
;
вектор п
является
осью симметрии гибридной орбитали (рис.
1). При изменении ориентации в пространстве
осей координат коэффициенты линейной
комбинации могут изменяться, однако
остается постоянным отношение сумм
квадратов коэффициентов для данного
значения азимутального квантового
числа. Это отношение определяет тип
гибридной орбитали.
Например,
орбиталь
 относится
к типу sapb,
где
а
и
b-
числа,
подобранные так, чтобы
относится
к типу sapb,
где
а
и
b-
числа,
подобранные так, чтобы
 .
Обычно принимают а
— 1,
b
=
1, 2 или 3.
,/p>
.
Обычно принимают а
— 1,
b
=
1, 2 или 3.
,/p>
Как правило, гибридные орбитали данного атома в молекуле относятся к одному типу, который называют типом гибридизации атома. Так, атом N в молекуле аммиака имеет гибридизацию атомных орбиталей типа sp3, атом С в молекуле этилена - sp2-гибридизацию, атом С в молекуле ацетилена - sp-гибридизацию. Это дает основание отождествлять тип гибридизации атома в молекуле с символом его некоей гипотетической электронной конфигурации.
Обычно система гибридных орбиталей строится таким образом, чтобы для разных орбиталей одногибридная орбиталь атома интегралы перекрывания были равны нулю. Кроме того, каждая орбиталь в молекуле либо остается негибридизированной атомной орбиталью, либо выбирается гибридизированной по определенному типу. Этим требованиям удовлетворяют несколько наборов орбиталей. Например, атом О в молекуле Н2О можно считать как sp2-, так и 5р3-гибридизированным.
Оси симметрии всех орбиталей обычно образуют симметричную фигуру (см. рис. 2). Операции симметрии этой фигуры переводят гибридные орбитали одного атома друг в друга. Такие гибридные орбитали называют эквивалентными. Например, линейная комбинация четырех валентных атомных орбиталей атома С приводит к четырем 5р3-гибридным орбиталям, оси симметрии которых по отношению друг к другу расположены под углом 109,5°, т.е. направлены по углам тетраэдра (рис. 2, в).

Отклонение конфигурации молекулы от симметричной связывают с взаимодействием химических связей (напр., с отталкиванием пар электронов, образующих связь). В такой форме представления о гибридизации атомных орбиталей используются в стереохимии.
Sp у BeH2 sp2 у BH3 sp3 у остальных
18) Ионная связь. Водородная связь. Внутримолекулярная и межмолекулярная водородная связь.
Ионная связь
Связь такого типа осуществляется в результате взаимного электростатического притяжения противоположно заряженных ионов. Ионы могут быть простыми, т. е. состоящими из одного атома (например Na+, K+, F-, С1-), или сложными, т. е. состоящими из двух или более атомов (например NH4+, ОН-, NO3-, SO42-). Простые ионы, обладающие положительным зарядом, легче всего образуются из атомов элементов с низким потенциалом ионизации; к таким элементам относятся металлы главных подгрупп I и II группы. Образование простых отрицательно заряженных ионов, напротив, характерно для атомов типичных неметаллов, обладающих большим сродством к электрону. Поэтому к типичным соединениям с ионным типом связи относятся галогениды щелочных металлов, например: NaCl, CsF и т. п.
В отличие от ковалентной связи, ионная связь не обладает направленностью. Это объясняется тем, что электрическое поле иона обладает сферической симметрией, т. е. убывает с расстоянием по одному и тому же закону в любом направлении. Поэтому взаимодействие между ионами осуществляется одинаково независимо от направления. Отсутствие у ионной связи направленности и насыщаемости обусловливает склонность ионных молекул к ассоциации, т. е. к соединению их друг с другом.
Водородная связь
Водородная связь — форма ассоциации между электроотрицательным атомом и атомом водорода H, связанным ковалентно с другим электроотрицательным атомом. В качестве электроотрицательных атомов могут выступать N, O или F. Водородные связи могут быть межмолекулярными или внутримолекулярными.
Часто водородную связь рассматривают как электростатическое взаимодействие, усиленное небольшим размером водорода, которое разрешает близость взаимодействующих диполей. Тогда об этом говорят как о разновидности донорно-акцепторной связи, невалентном взаимодействии между атомом водорода H, ковалентно связанным с атомом A группы A-H молекулы RA-H и электроотрицательным атомом B другой молекулы (или функциональной группы той же молекулы) BR'. Результатом таких взаимодействий являются комплексы RA-H•••BR' различной степени стабильности, в которых атом водорода выступает в роли «моста», связывающего фрагменты RA и BR'.
Особенностями водородной связи, по которым её выделяют в отдельный вид, является её не очень высокая прочность, её распространенность и важность, особенно в органических соединениях, а также некоторые побочные эффекты, связанные с малыми размерами и отсутствием дополнительных электронов у водорода.
В настоящее время в рамках теории молекулярных орбиталей водородная связь рассматривается как частный случай ковалентной с делокализацией электронной плотности по цепи атомов и образованием трёхцентровых четырёхэлектронных связей (например, -H•••[F-H•••F]-).
Межмолекулярная и внутримолекулярная водородная связь
Водородные связи обнаружены во многих химических соединениях. Они возникают, как правило, между атомами фтора, азота и кислорода (наиболее электроотрицательные элементы), реже - при участии атомов хлора, серы и других неметаллов. Прочные водородные связи образуются в таких жидких веществах, как вода, фтороводород, кислородсодержащие неорганические кислоты, карбоновые кислоты, фенолы, спирты, аммиак, амины. При кристаллизации водородные связи в этих веществах обычно сохраняются. Поэтому их кристаллические структуры имеют вид цепей (метанол), плоских двухмерных слоев (борная кислота), пространственных трехмерных сеток (лед).
Если водородная связь объединяет части одной молекулы, то говорят о внутримолекулярной водородной связи. Это особенно характерно для многих органических соединений . Если же водородная связь образуется между атомом водорода одной молекулы и атомом неметалла другой молекулы (межмолекулярная водородная связь), то молекулы образуют довольно прочные пары, цепочки, кольца. Так, муравьиная кислота и в жидком и в газообразном состоянии существует в виде димеров: а газообразный фтороводород содержат полимерные молекулы, включающие до четырех частиц HF. Прочные связи между молекулами можно найти в воде, жидком аммиаке, спиртах. Необходимые для образования водородных связей атомы кислорода и азота содержат все углеводы, белки, нуклеиновые кислоты. Известно, например, что глюкоза, фруктоза и сахароза прекрасно растворимы в воде. Не последнюю роль в этом играют водородные связи, образующиеся в растворе между молекулами воды и многочисленными OH-группами углеводов.
19) Диаграмма состояния воды. Правило фаз Гиббса-Коновалова и его применение для характеристики однокомпонентной системы.
Диаграмма состояния воды — система с одним компонентом H2O, поэтому наибольшее число фаз, которые одновременно могут находиться в равновесии, равно трем. Эти три фазы — жидкость, лед, пар. Число степеней свободы в этом случае равно нулю, т.е. нельзя изменить ни давление, ни температуру, чтобы не исчезла ни одна из фаз. Обычный лед, жидкая вода и водяной пар могут существовать в равновесии одновременно только при давлении 0,61 кПа и температуре 0,0075°С. Точка сосуществования трех фаз называется тройной точкой (O).

Кривая ОС разделяет области пара и жидкости и представляет собой зависимость давления насыщенного водяного пара от температуры. Кривая ОС показывает те взаимосвязанные значения температуры и давления, при которых жидкая вода и водяной пар находятся в равновесии друг с другом, поэтому она называется кривой равновесия жидкость—пар или кривой кипения.
Кривая ОВ отделяет область жидкости от области льда. Она является кривой равновесия твердое состояние—жидкость и называется кривой плавления. Эта кривая показывает те взаимосвязанные пары значений температуры и давления, при которых лед и жидкая вода находятся в равновесии.
Кривая OA называется кривой сублимации и показывает взаимосвязанные пары значений давления и температуры, при которых в равновесии находятся лед и водяной пар.
Правило фаз (уравнение) Гиббса, известное также как закон фазового равновесия, было сформулировано в 1876 г. Оно относится к одному из общих законов физики и химии и является выражением второго закона термодинамики применительно к фазовым равновесиям. С его помощью можно описать и гомогенные и гетерогенные химические равновесия, а именно: -термодинамически установить (предсказать) возможность или невозможность существования данной гомогенной или гетерогенной системы; -вычислить максимальное число фаз, которые одновременно могут находиться в равновесии при данных условиях; -априори характеризовать фазовый состав сколь угодно сложной многокомпонентной системы при заданном числе независимых переменных, определяющих равновесие; -предсказать по числу степеней свободы поведение исследуемой системы, т.е. установить варианты возможных фазовых превращений при изменении одного, двух и более внешних условий (при сохранении остальных параметров состояния системы постоянными).
В общем виде правило фаз Гиббса формулируется следующим образом: число термодинамических степеней свободы, или вариантность равновесной термодинамической системы С, определяется как разность числа независимых компонентов системы К и числа сосуществующих фаз Ф плюс число внешних факторов n, влияющих на равновесие. Его математическое выражение записывается в виде простого соотношения: С = К – Ф + n. Анализируя это выражение можно сделать следующие выводы: -число степеней свободы равновесной системы возрастает с увеличением числа независимых компонентов и понижается с увеличением числа сосуществующих фаз; - число степеней свободы может быть равно либо нулю, либо целому положительному числу, т.е. С³0; - так как число степеней свободы не может быть отрицательным, то число сосуществующих фаз в равновесной термодинамической системе в общем случае не может превышать суммы К + n.
Другая запись

где j — число фаз (например, агрегатных состояний вещества);
v — число степеней свободы, то есть независимых параметров (температура, давление, концентрация компонентов), которые полностью определяют состояние системы при равновесии и которые можно менять без изменения числа и природы фаз;
k — число компонентов системы — число входящих в систему индивидуальных веществ за вычетом числа химических уравнений, связывающих эти вещества. Иначе говоря, это минимальное количество веществ, из которых можно приготовить каждую фазу системы.
n — число переменных, характеризующих влияние внешних условий на равновесие системы.
В случае однокомпонентной системы оно упрощается до:

Отсюда видно, например, что в однокомпонентной системе три фазы (j=3) могут сосуществовать при числе степеней свободы v, равном нулю, то есть при фиксированных давлении и температуре, что соответствует тройной точке на фазовой диаграмме. Две фазы (j=2) сосуществуют при произвольном изменении либо давления, либо температуры, когда вторая из этих переменных не является независимой (v=1), то есть двухфазному равновесию на фазовой диаграмме соответствует линия. Если фаза одна (j=1), число степеней свободы системы равно двум, то есть температура и давление могут менятся независимо в пределах некоторой области на фазовой диаграмме — пока система не окажется на одной из линий двухфазного равновесия.
20) Скорость химической реакции. Факторы, влияющие на скорость. Закон действующих масс. Константа скорости. Молекулярность и порядок реакции.
Скорость химической реакции — изменение количества вещества одного из реагирующих веществ за единицу времени в единице реакционного пространства. Является ключевым понятием химической кинетики. Скорость химической реакции — величина всегда положительная, поэтому, если она определяется по исходному веществу (концентрация которого убывает в процессе реакции), то полученное значение домножается на −1.
Например для реакции:

выражение для скорости будет выглядеть так:
 .
.
В 1865 году Н. Н. Бекетовым и в 1867 году Гульдбергом и Вааге был сформулирован закон действующих масс:
Скорость химической реакции в каждый момент времени пропорциональна концентрациям реагентов, возведенным в некоторые степени.
Для элементарных реакций показатель степени при значении концентрации каждого вещества часто равен его стехиометрическому коэффициенту, для сложных реакций это правило не соблюдается. Кроме концентрации на скорость химической реакции оказывают влияние следующие факторы:
природа реагирующих веществ,
наличие катализатора,
температура (правило Вант-Гоффа),
давление,
площадь поверхности реагирующих веществ.
Если мы рассмотрим самую простую химическую реакцию A + B → C, то мы заметим, что мгновенная скорость химической реакции величина непостоянная.
Константа скорости реакции (удельная скорость реакции) — коэффициент пропорциональности в кинетическом уравнении.
Физический смысл константы скорости реакции k следует из уравнения закона действующих масс: k численно равна скорости реакции при концентрации каждого из реагирующих веществ равной 1 моль/л.
Константа скорости реакции зависит от температуры, от природы реагирующих веществ, но не зависит от их концентрации.
Порядок реакции по данному веществу — показатель степени при концентрации этого вещества в кинетическом уравнении реакции.
Реакция нулевого порядка
График зависимости концентрации реагента A в реакции A → B от времени для нулевого порядка реакции
Кинетическое уравнение имеет следующий вид:
V0 = k0
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ. Нулевой порядок характерен, например, для гетерогенных реакций в том случае, если скорость диффузии реагентов к поверхности раздела фаз меньше скорости их химического превращения.
Реакция первого порядка
График зависимости концентрации реагента A для первого порядка реакции
Кинетическое уравнение реакции первого порядка:

Константа скорости реакции вычисляется как тангенс угла наклона прямой к оси времени:
k1 = − tgα
Реакция второго порядка
График зависимости концентрации реагента A для второго порядка реакции
Для реакций второго порядка кинетическое уравнение имеет следующий вид:

Константа скорости реакции равна тангенсу угла наклона прямой к оси времени:
k2 = − tgα