

Micro-Nano Technology for Genomics and Proteomics BioMEMs - Ozkan
.pdf132 |
JOANNA S. ALBALA |
residues 79–376, and this region of Rad51C also interacts with Rad51D and Xrcc3. We have also determined that the N-terminal domain of Rad51D, amino acid residues 4–77, binds to Xrcc2 while the C-terminal domain of Rad51D, amino acid residues 77–328, binds Rad51C [50].
5.3.2. Protein Interaction Arrays
We have generated protein interaction microarrays to examine protein-protein and protein-DNA interactions for the Rad51 paralogs. Protein microarrays were generated to contain various DNA repair proteins, histones, nucleosomes, various antibodies and DNA. We applied these various proteins, antibodies and DNA to functionalized glass slides and interrogated the slides with Rad51B and Rad51C to identify novel protein-protein interactions for proteins involved in DNA double-strand break repair.
Standard microarray spotting techniques were used to attach proteins to glass slides in a microarray format to analyze protein-protein interactions. Proteins were resuspended in spotting buffer and arrayed in duplicate on aminopropyl triethoxysilane and/or poly- lysine-coated glass slides using a robotic arrayer. Approximately two hundred proteins were spotted on the array. Scanning and analysis of the arrays were performed on a ScanArray 5000 (488 nm laser for FITC scans and 543 nm laser for rhodamine) using QuantArray software. All information regarding the construction and use of the protein arrays can be found at http://bbrp.llnl.gov/microarrays/external/index.html.
We have shown that the DNA repair protein RAD51B, and not its cognate partner RAD51C, interacts with histones and not nucleosomes (see Figure 5.2). Several proteins
A. |
B. |
FIGURE 5.2. Protein interaction microarrays show that Rad51B interacts with histones and not nucleosomes. Protein arrays were generated to contain a variety of DNA repair proteins, histones, nucleosomes and antibodies. Proteins were generated in a miniaturized baculoviral expression system or as GFP fusion proteins by cell-free expression in E. coli. Panel A: Protein interaction microarray incubated without recombinant Rad51B. Panel B: Protein interaction microarray incubated with recombinant Rad51B protein. The two red spots indicate an interaction between Rad51B and histones on the array. No interaction is observed between Rad51B and the adjacent nucleosome proteins. Green spots are GFP fusion proteins. Red spots in the top row are rhodamine-labeled DNA markers for addressability of spots on the array. Two hundred and forty-two spots were robotically arrayed and only a fraction of these are shown.
HITTING THE SPOT: THE PROMISE OF PROTEIN MICROARRAYS |
133 |
on this array incorporated GFP into the final fusion protein [63]. Unique RAD51B-histone interactions were corroborated using Far Western analysis [51]. This is the first demonstration of an interaction between RAD51B and histone proteins that may be important for the successful repair of DNA double-strand breaks.
5.4. SUMMARY: PROTEIN ARRAYS-HOPE OR HYPE?
Protein microarrays hold great promise for accelerating basic biology, biomarker identification and drug discovery. Recent strides have been made in the use of protein arrays for diagnosis of autoimmune disorders [21, 31, 64, 65] as well as for cancer biomarker identification [66]. Of particular note is the use of reverse arrays pioneered by Petricion and Liotta where subpopulations of cells from a given tissue were obtained by use of laser capture microdissection, cell lysates were made, and the lysates interrogated for biomarker detection on arrays [67]. Another approach relies on the identification of panel of protein biomarkers or signatures for diagnosis or prognosis of disease [68].
There is still much to be learned in the protein array arena, particularly in standardization of array production and content for comparative analysis of results between labs. Future work will aim to utilize protein microarrays to understand complex subproteomes and the characterization of posttranslational modification of proteins. Although the work ahead is challenging, the payoff for miniaturized, high-throughput analysis for personalized medicine is enormous. Progress has been made, but the best may yet be to come.
ACKNOWLEDGEMENTS
Thanks to Kristi Miller, Daniel Yoshikawa, Peter Beernick and Matthew Coleman for their contribution to the production of the proteins and protein interaction microarrays. The author would like to thank Marianne Kavanagh, Irene Jones and Christa Prange for critical review of this manuscript. This work was performed under the auspices of the U.S. Department of Energy managed by the University of California-Lawrence Livermore National Laboratory under Contract No. W-7405-Eng-48.
REFERENCES
[1]B.B. Haab, M.J. Dunham, and P.O. Brown. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome. Biol., 2:RESEARCH0004, 2001.
[2]H. Zhu, J.F. Klemic, S. Chang, P. Bertone, A. Casamayor, K.G. Klemic, D. Smith, M. Gerstein, M.A. Reed, and M. Snyder. Analysis of yeast protein kinases using protein chips. Nat. Genet., 26:283–289, 2000.
[3]J.S. Albala. Array-based proteomics: the latest chip challenge. Expert. Rev. Mol. Diagn., 1:145–152, 2001.
[4]D.J. Cahill. Protein and antibody arrays and their medical applications. J. Immunol. Methods, 250:81–91, 2001.
[5]B. Schweitzer, P. Predki, and M. Snyder. Microarrays to characterize protein interactions on a whole-proteome scale. Proteomics, 3:2190–2199, 2003.
[6]H. Zhu, M. Bilgin, R. Bangham, D. Hall, A. Casamayor, P. Bertone, N. Lan, R. Jansen, S. Bidlingmaier, and T. Houfek et al. Global analysis of protein activities using proteome chips. Science, 293:2101–2105, 2001.
[7]J. Ziauddin and D.M. Sabatini. Microarrays of cells expressing defined cDNAs. Nature, 411:107–110, 2001.
134 |
JOANNA S. ALBALA |
[8]J. Kononen, L. Bubendorf, A. Kallioniemi, M. Barlund, P. Schraml, S. Leighton, J. Torhorst, M.J. Mihatsch, G. Sauter, and O.P. Kallioniemi. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med., 4:844–847, 1998.
[9]G. Lennon, C. Auffray, M. Polymeropoulos, and M.B. Soares. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics, 33:151–152, 1996.
[10]R.L. Strausberg, E.A. Feingold, R.D. Klausner, and F.S. Collins. The mammalian gene collection. Science, 286:455–457, 1999.
[11]L. Brizuela, P. Braun, and J. LaBaer. FLEXGene repository: from sequenced genomes to gene repositories for high-throughput functional biology and proteomics. Mol. Biochem. Parasitol., 118:155–165, 2001.
[12]M. Gilbert and J.S. Albala. Accelerating code to function: sizing up the protein production line. Curr. Opin. Chem. Biol., 6:102–105, 2002.
[13]S.A. Lesley, P. Kuhn, A. Godzik, A.M. Deacon, I. Mathews, A. Kreusch, G. Spraggon, H.E. Klock, D. McMullan, and T. Shin et al. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. U.S.A., 99:11664–11669, 2002.
[14]R.Y. Huang, S.J. Boulton, M. Vidal, S.C. Almo, A.R. Bresnick, and M.R. Chance. High-throughput expression, purification, and characterization of recombinant Caenorhabditis elegans proteins. Biochem. Biophys. Res. Commun., 307:928–934, 2003.
[15]P. Braun, Y. Hu, B. Shen, A. Halleck, M. Koundinya, E. Harlow, and J. LaBaer. Proteome-scale purification of human proteins from bacteria. Proc. Natl. Acad. Sci. U.S.A., 99:2654–2659, 2002.
[16]P. Braun and J. LaBaer. High throughput protein production for functional proteomics. Trends Biotechnol., 21:383–388, 2003.
[17]P. Sebastian, J. Wallwitz, and S. Schmidt. Semi automated production of a set of different recombinant GST-Streptag fusion proteins. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 786:343–355, 2003.
[18]Y.P. Shih, W.M. Kung, J.C. Chen, C.H. Yeh, A.H. Wang, and T.F. Wang. High-throughput screening of soluble recombinant proteins. Protein Sci., 11:1714–1719, 2002.
[19]D. Busso, R. Kim, and S.H. Kim. Expression of soluble recombinant proteins in a cell-free system using a 96-well format. J. Biochem. Biophys. Methods, 55:233–240, 2003.
[20]L.J. Holt, K. Bussow, G. Walter, and I.M. Tomlinson. By-passing selection: direct screening for antibodyantigen interactions using protein arrays. Nucleic Acids Res., 28:E72, 2000.
[21]A. Lueking, A. Possling, O. Huber, A. Beveridge, M. Horn, H. Eickhoff, J. Schuchardt, H. Lehrach, and D.J. Cahill. A Nonredundant Human Protein Chip for Antibody Screening and Serum Profiling. Mol. Cell. Proteomics, 2:1342–1349, 2003.
[22]P. Angenendt, J. Glokler, Z. Konthur, H. Lehrach, and D.J. Cahill. 3D protein microarrays: performing multiplex immunoassays on a single chip. Anal. Chem., 75:4368–4372, 2003.
[23]N. Stich, G. van Steen, and T. Schalkhammer. Design and peptide-based validation of phage display antibodies for proteomic biochips. Comb. Chem. High Throughput Screen, 6:67–78, 2003.
[24]H.J. de Haard, N. van Neer, A. Reurs, S.E. Hufton, E.C. Roovers, P. Henderikx, A.P. de Bruine, J.W. Arends, and H.R. Hoogenboom. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem., 274:18218–18230, 1999.
[25]S. Weng, K. Gu, P.W. Hammond, P. Lohse, C. Rise, R.W. Wagner, M.C. Wright, and R.G. Kuimelis. Generating addressable protein microarrays with PROfusion covalent mRNA-protein fusion technology. Proteomics, 2:48–57, 2002.
[26]H. Petach and L. Gold. Dimensionality is the issue: use of photoaptamers in protein microarrays. Curr. Opin. Biotechnol., 13:309–314, 2002.
[27]E.N. Brody and L. Gold. Aptamers as therapeutic and diagnostic agents. J. Biotechnol., 74:5–13, 2000.
[28]Y. Lin, R. Huang, X. Cao, S.M. Wang, Q. Shi, and R.P. Huang. Detection of multiple cytokines by protein arrays from cell lysate and tissue lysate. Clin. Chem. Lab. Med., 41:139–145, 2003.
[29]G. MacBeath and S.L. Schreiber. Printing proteins as microarrays for high-throughput function determination. Science, 289:1760–1763, 2000.
[30]P. Angenendt, J. Glokler, D. Murphy, H. Lehrach, and D.J. Cahill. Toward optimized antibody microarrays: a comparison of current microarray support materials. Anal. Biochem., 309:253–260, 2002.
[31]T.O. Joos, M. Schrenk, P. Hopfl, K. Kroger, U. Chowdhury, D. Stoll, D. Schorner, M. Durr, K. Herick, and S. Rupp et al. A microarray enzyme-linked immunosorbent assay for autoimmune diagnostics. Electrophoresis, 21:2641–2650, 2000.
HITTING THE SPOT: THE PROMISE OF PROTEIN MICROARRAYS |
135 |
[32]L.G. Mendoza, P. McQuary, A. Mongan, R. Gangadharan, S. Brignac, and M. Eggers. High-throughput microarray-based enzyme-linked immunosorbent assay (ELISA). Biotechniques, 27:778–780, 782–776, 788, 1999.
[33]V. Afanassiev, V. Hanemann, and S. Wolfl. Preparation of DNA and protein micro arrays on glass slides coated with an agarose film. Nucleic Acids Res., 28:E66, 2000.
[34]D. Guschin, G. Yershov, A. Zaslavsky, A. Gemmell, V. Shick, D. Proudnikov, P. Arenkov, and A. Mirzabekov. Manual manufacturing of oligonucleotide, DNA, and protein microchips. Anal. Biochem., 250:203–211, 1997.
[35]J.C. Miller, H. Zhou, J. Kwekel, R. Cavallo, J. Burke, E.B. Butler, B.S. Teh, and B.B. Haab. Antibody microarray profiling of human prostate cancer sera: antibody screening and identification of potential biomarkers. Proteomics, 3:56–63, 2003.
[36]J. Madoz-Gurpide, H. Wang, D.E. Misek, F. Brichory, and S.M. Hanash. Protein based microarrays: a tool for probing the proteome of cancer cells and tissues. Proteomics, 1:1279–1287, 2001.
[37]J. Turkova. Oriented immobilization of biologically active proteins as a tool for revealing protein interactions and function. J. Chromatogr. B Biomed. Sci. Appl., 722:11–31, 1999.
[38]P. Pavlickova, A. Knappik, D. Kambhampati, F. Ortigao, and H. Hug. Microarray of recombinant antibodies using a streptavidin sensor surface self-assembled onto a gold layer. Biotechniques, 34:124–130, 2003.
[39]P. Peluso, D.S. Wilson, D. Do, H. Tran, M. Venkatasubbaiah, D. Quincy, B. Heidecker, K. Poindexter,
N.Tolani, M. Phelan et al. Optimizing antibody immobilization strategies for the construction of protein microarrays. Anal. Biochem., 312:113–124, 2003.
[40]A. Lueking, M. Horn, H. Eickhoff, K. Bussow, H. Lehrach, and G. Walter. Protein microarrays for gene expression and antibody screening. Anal. Biochem., 270:103–111, 1999.
[41]J.F. Mooney, A.J. Hunt, J.R. McIntosh, C.A. Liberko, D.M. Walba, and C.T. Rogers. Patterning of functional antibodies and other proteins by photolithography of silane monolayers. Proc. Natl. Acad. Sci. U.S.A., 93:12287–12291, 1996.
[42]V.W. Jones, J.R. Kenseth, M.D. Porter, C.L. Mosher, and E. Henderson. Microminiaturized immunoassays using atomic force microscopy and compositionally patterned antigen arrays. Anal. Chem., 70:1233–1241, 1998.
[43]A. Roda, M. Guardigli, C. Russo, P. Pasini, and M. Baraldini. Protein microdeposition using a conventional ink-jet printer. Biotechniques, 28:492–496, 2000.
[44]J.B. Delehanty and F.S. Ligler. Method for printing functional protein microarrays. Biotechniques, 34:380– 385, 2003.
[45]D.S. Wilson and S. Nock. Recent developments in protein microarray technology. Angew. Chem. Int. Ed. Engl., 42:494–500, 2003.
[46]T.O. Joos, D. Stoll, and M.F. Templin. Miniaturised multiplexed immunoassays. Curr. Opin. Chem. Biol., 6:76–80, 2002.
[47]B. Schweitzer, S. Wiltshire, J. Lambert, S. O’Malley, K. Kukanskis, Z. Zhu, S.F. Kingsmore, P.M. Lizardi, and D.C. Ward. Inaugural article: immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc. Natl. Acad. Sci. U.S.A., 97:10113–10119, 2000.
[48]B. Schweitzer, S. Roberts, B. Grimwade, W. Shao, M. Wang, Q. Fu, Q. Shu, I. Laroche, Z. Zhou, and V.T. Tchernev et al. Multiplexed protein profiling on microarrays by rolling-circle amplification. Nat. Biotechnol., 20:359–365, 2002.
[49]J.S. Albala, K. Franke, I.R. McConnell, K.L. Pak, P.A. Folta, B. Rubinfeld, A.H. Davies, G.G. Lennon, and
R.Clark. From genes to proteins: high-throughput expression and purification of the human proteome. J. Cell. Biochem., 80:187–191, 2000.
[50]K.A. Miller, D. Sawicka, D. Barsky, and J.S. Albala. Domain mapping of the Rad51 paralog protein complexes. Nucleic Acids Res., 32:169–178, 2004.
[51]M.A. Coleman, K.A. Miller, P.T. Beernink, D.M. Yoshikawa, and J.S. Albala. Identification of chromatinrelated protein interactions using protein microarrays. Proteomics, 3:2101–2107, 2003.
[52]L.H. Thompson and D. Schild. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res., 477:131–153, 2001.
[53]S.C. West. Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell. Biol., 4:435–445, 2003.
[54]D.L. Pittman, L.R. Weinberg, and J.C. Schimenti. Identification, characterization, and genetic mapping of Rad51d, a new mouse and human RAD51/RecA-related gene. Genomics, 49:103–111, 1998.
136 |
JOANNA S. ALBALA |
[55]N. Liu, J.E. Lamerdin, R.S. Tebbs, D. Schild, J.D. Tucker, M.R. Shen, K.W. Brookman, M.J. Siciliano, C.A. Walter, and W. Fan et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol. Cell., 1:783–793, 1998.
[56]M.K. Dosanjh, D.W. Collins, W. Fan, G.G. Lennon, J.S. Albala, Z. Shen, and D. Schild. Isolation and characterization of RAD51C, a new human member of the RAD51 family of related genes. Nucleic Acids Res., 26:1179–1184, 1998.
[57]J.S. Albala, M.P. Thelen, C. Prange, W. Fan, M. Christensen, L.H. Thompson, and G.G. Lennon. Identification of a novel human RAD51 homolog, RAD51B. Genomics, 46:476–479, 1997.
[58]N. Liu, D. Schild, M.P. Thelen, and L.H. Thompson. Involvement of Rad51C in two distinct protein complexes of Rad51 paralogs in human cells. Nucleic Acids Res., 30:1009–1015, 2002.
[59]J.Y. Masson, M.C. Tarsounas, A.Z. Stasiak, A. Stasiak, R. Shah, M.J. McIlwraith, E.E. Benson, and S.C. West. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev., 15:3296–3307, 2001.
[60]K.A. Miller, D.M. Yoshikawa, I.R. McConnell, R. Clark, D. Schild, and J.S. Albala. RAD51C Interacts with RAD51B and Is Central to a Larger Protein Complex in Vivo Exclusive of RAD51. J. Biol. Chem., 277:8406–8411, 2002.
[61]C. Wiese, D.W. Collins, J.S. Albala, L.H. Thompson, A. Kronenberg, and D. Schild. Interactions involving the Rad51 paralogs Rad51C and XRCC3 in human cells. Nucleic Acids Res., 30:1001–1008, 2002.
[62]D.S. Shin, L. Pellegrini, D.S. Daniels, B. Yelent, L. Craig, D. Bates, D.S. Yu, M.K. Shivji, C. Hitomi, A.S. Arvai, N. Volkmann, H. Tsuruta, T.L. Blundell, A.R. Venkitaraman, and J.A. Tainer. Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J., 22:4566– 4576, 2003.
[63]P.T. Beernink, S.S. Krupka, V. Lao, G. Martin, and M.A. Coleman. Application of in vitro protein expression to human prote. Sci. World J., 2:73–74, 2002.
[64]W.H. Robinson, L. Steinman, and P.J. Utz. Protein arrays for autoantibody profiling and fine-specificity mapping. Proteomics, 3:2077–2084, 2003.
[65]W.H. Robinson, C. DiGennaro, W. Hueber, B.B. Haab, M. Kamachi, E.J. Dean, S. Fournel, D. Fong, M.C. Genovese, and H.E. de Vegvar et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat. Med., 8:295–301, 2002.
[66]J.C. Miller, E.B. Butler, B.S. Teh, and B.B. Haab. The application of protein microarrays to serum diagnostics: prostate cancer as a test case. Dis. Markers, 17:225–234, 2001.
[67]R.L. Grubb, V.S. Calvert, J.D. Wulkuhle, C.P. Paweletz, W.M. Linehan, J.L. Phillips, R. Chuaqui, A. Valasco, J. Gillespie, and M. Emmert-Buck et al. Signal pathway profiling of prostate cancer using reverse phase protein arrays. Proteomics, 3:2142–2146, 2003.
[68]E.F. Petricoin, A.M. Ardekani, B.A. Hitt, P.J. Levine, V.A. Fusaro, S.M. Steinberg, G.B. Mills, C. Simone, D.A. Fishman, and E.C. Kohn et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet, 359:572–577, 2002.
6
Use of Electric Field Array Devices for Assisted Assembly of DNA Nanocomponents and Other Nanofabrication Applications
Michael J. Heller1, Cengiz S. Ozkan2, and Mihrimah Ozkan3
1University of California San Diego, La Jolla, CA 92093, and Nanogen, San Diego, CA, 92121
2Department of Mechanical Engineering, University of California Riverside, Riverside CA 92521 3Department of Electrical Engineering, University of California Riverside, Riverside CA 92521
Microelectronic arrays utilizing electric field transport have been developed for DNA diagnostics (including infectious and genetic disease and cancer detection), for short tandem repeat (STR) forensics analysis, and for gene expression applications. In addition to these bioresearch and clinical diagnostic applications, such devices also have the potential to carry out the assisted assembly of a wide variety of molecular scale, nanoscale and microscale components into higher order structures. These microelectronic array devices are able to produce defined electric fields on their surfaces that allow molecules and other entities with high fidelity recognition properties to be transported to or from any site on the surface of the array. Such devices can utilize either DC electric fields which cause movement of entities by their relative charge, or AC electric fields which allow entities to be selectively positioned by their dielectric properties. An almost unlimited variety of molecules and nanocomponents can be utilized with these devices, including: DNA, DNA constructs with fluorescent, photonic or electronic transfer properties, RNA, RNA constructs, amino acids, peptides, proteins (antibodies, enzymes), nanoparticles (quantum dots, carbon nanotubes, nanowires), cells and even micron scale semiconductor components. Thus, electric field devices can be used for developing a unique highly parallel “Pick & Place” fabrication process by which a variety of heterogeneous molecules, nanocomponents and micron sized
138 |
MICHAEL J. HELLER, CENGIZ S. OZKAN AND MIHRIMAH OZKAN |
objects with intrinsic self-assembly properties can be organized into higher order 2D and 3D structures and devices. The process represents a unique synergy of combining the best aspects of a “top-down” process with a “bottom-up” process. Finally, integration of optical tweezers for manipulation of live cells and microspheres in a similar microarray setup is demonstrated for the applications of biological delivery and invasive manipulation of these species.
6.1. INTRODUCTION
Nanotechnology and nanoscience encompass a wide range of new concepts, ideas and potential applications for nanoelectronics, novel materials, more efficient energy conversion processes, and a new generation of sensors and biomedical devices [1]. Generally, molecular or nanoelectronic devices are envisioned as the more revolutionary outcome of this new field. Presently, there are a numerous examples of novel nanocomponents (organic electron transfer molecules, quantum dots, carbon nanotubes, nanowires, etc.), and some examples of a first level assembly of these nanocomponents into simple structures with some higher order properties [2–4]. Nevertheless, the larger issue of developing viable nanofabrication techniques that allow billions of molecular/nanoscale components to be assembled and interconnected into useful logic/memory devices still remains a considerable challenge. The one exception to this is the present silicon/CMOS fabrication technology which continues to shrink semiconductor feature sizes down to the nanoscale level, although it does appear that this photolithographic based “top-down” technique is being pressed to it’s limit. In addition to the nanoelectronic applications, other new nanodevices and nanomaterials with higher order photonic, mechanical, mechanistic, sensory, chemical, catalytic, and therapeutic properties are also envisioned [1]. Again, a key problem in enabling such new devices and materials will most likely be in developing viable highly parallel fabrication technologies for organizing and integrating heterogeneous components of different sizes and compositions into higher level structures and devices.
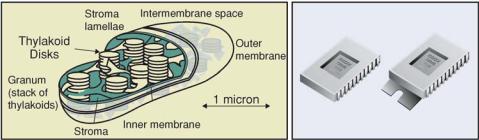
Biology provides some of the best examples of nanotechnology in action, and biological self-assembly or self-organization might be considered the primer for developing so-called “bottom-up” processes for nanofabrication. Our present knowledge of the molecular biology of living systems demonstrates many unique molecular scale nanostructures and mechanisms which include efficient electron transfer and photonic transfer systems, energy conversion systems, high information content molecules (DNA and RNA), precision structural components (proteins, fatty acids, etc) and a wide variety of highly efficient chemomechanical (catalytic) molecules called enzymes. By way of example, in the photosynthetic energy conversion systems of plants and algae, the chloroplasts contain solid state antenna structures (50–100 nanometers) that are composed of hundreds of highly organized chromophore molecules and other nanostructures. These nanoscale antennas are able to collect photonic energy and transfer it to other structures with very high quantum efficiency. The process is quite different than the way a man-made device, such as charged coupled device (CCD) would capture photons and convert them into electrical energy (Figure 6.1). To date, it has not been possible to design a biomimetic model of these solid state antenna structures that retains any of the efficient properties of the biological systems. In some sense, the problem with enabling this type nanotechnology is like trying to implement
USE OF ELECTRIC FIELD ARRAY DEVICES FOR ASSISTED ASSEMBLY |
139 |
FIGURE 6.1. Comparison of one of nature’s photonic conversion devices, a plant chloroplast, with a man-made charged coupled device (CCD). The chloroplast is a highly efficient structure which contains organized antenna structures that capture light energy and transfer it via the Forster resonant energy transfer process to one single molecule, which then converts the resonant energy into chemical energy. The CCD device also captures photons and converts them to an electrical signal. The overall efficiency of the chloroplast energy conversion process is much higher than for any man made device.
the microelectronics revolution with a basic understanding of semiconductor properties, but without the photolithography fabrication process being available to carry out the integration of components into the higher order structures. Living systems on the other hand have developed the ultimate “bottom-up” processes that allows component molecules and nanostructures with intrinsic self-assembly properties to be synthesized, and then further organized into more intricate three dimensional nanostructures, organelles, cells and final organisms.
Of all the biological based molecules available with high recognition and self-assembly properties, the nucleic acids represent one of the more promising materials which may be useful for creating molecular electronic/photonic circuits, organized nanostructures, and even integrated microelectronic and photonic devices [5–7]. The nucleic acids, deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) are programmable molecules which, via their base sequence, have intrinsic molecular recognition and self-assembly properties. Short DNA sequences, called oligonucleotides, are easily synthesized and readily modified with a variety of functional groups. The ability to functionalize synthetic DNA molecules with fluorescent chromophore and charge transfer groups provides a means to incorporate electronic and photonic properties directly into these molecules. DNA can also be used to functionalize larger molecules, nanostructures (quantum dots, gold nanoparticles, carbon nanotubes, etc.) or even micron size components which then can self-assemble or be selectively attached to surfaces, including glass, silicon or other semiconductor materials. DNA molecules, in particular synthetic oligonucleotides represent an ideal type of “molecular legos” for self-assembly of nanocomponents into more complex two and three dimensional higher order structures. At a first level, DNA can be used for a kind of template directed assembly on solid surfaces. This technique involves taking complementary DNA strands and using them as a selective glue to bind other organic or inorganic structures together, or to surfaces. DNA molecules contain base moieties, cytosine, guanine, adenine, and thymine (C,G,A,T) which will only bind to each other in specific pairs: C with G, and A with T. This base pairing property allows single strands of DNA to recognize each other, and bind together to form double-stranded DNA structures. Consequently, a 5’-ATTTGC-3’
140 |
MICHAEL J. HELLER, CENGIZ S. OZKAN AND MIHRIMAH OZKAN |
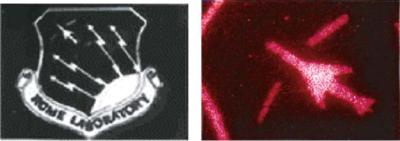
FIGURE 6.2. Shows two examples of DNA imprinting and patterning on a silicon substrate material. On the left, the logo was created by covering the silicon substrate with a DNA capture probe sequence, and then patterned using a mask and UV light to inactivate the exposed DNA sequences. The substrate was then hybridized with a fluorescent complementary DNA sequence, creating the fluorescent logo. The jet logo on the right was created by covering a silicon substrate with a DNA capture probe sequence and then patterned using a mask and UV light to inactivate the exposed DNA sequences. The substrate was then hybridized with fluorescent 200 nanometer nanospheres to which complementary DNA sequences were attached.
strand will only bind strongly with its complement 5’-GCAAAT-3” strand. DNA strands can be attached to surfaces (glass, silicon, gold, etc.) and then patterned by UV light exposure through a mask. Complementary DNA strands can be derivatized with fluorescent molecules or attached to nanostructures (quantum dots, fluorescent nanoparticles, metallic particles). When the specific fluorescent DNA or DNA nanostructures are hybridized to the complementary patterned DNA on a solid support, the patterned substrate material shows the fluorescent image. This DNA assembly method is best suited for the organization of small nanostructures on surfaces, but can be extended to structures in the micron size range. Figure 6.2 shows an example of UV patterned DNA silicon substrate hybridized with complementary fluorescent DNA sequences and with 200 nanometer fluorescent DNA nanospheres.
Active microelectronic arrays have been developed for a number of applications in bioresearch and DNA clinical diagnostics [8–17]. These active microelectronic devices have the ability to create almost any electric field transport geometry on the array surface, which then allows charged reagent and analyte molecules (DNA, RNA, proteins, enzymes), nanostructures, cells and micron-scale structures to be moved to or from any of the microscopic test sites on the device surface. When specific DNA hybridization reactions are carried out on the array, the device is leveraging the electric fields to direct the self-assembly of DNA molecules at the specified microlocation on the chip surface. Microelectronic arrays have been used to demonstrate the organization of complex fluorescent DNA molecular structures and mechanisms within selected microlocations on the array device. In principle these active microelectronic array devices are serving as “motherboards or hostboards” that can carry out the assisted self-assembly of DNA derivatized molecules, nanostructures or microscale structures into more complex two and three dimensional structures [18–25]. This electric field assisted assembly technique is a type of “Pick & Place” process that has potential applications from the nearer term heterogeneous integration of micron-scale photonic, microelectronic and MEMS devices; to the development of high density data storage devices; to the longer term nanofabrication of true molecular and nanoelectronic circuits and devices (Figure 6.3).

USE OF ELECTRIC FIELD ARRAY DEVICES FOR ASSISTED ASSEMBLY |
141 |
Synergy of Bottom-Up and Top-Down Processes |
||||
Integratedt t Photonict ic |
|
Meso-Scalele |
|
|
Devicesi |
|
|
||
|
Heterogeneoust |
Integrationt ti |
||
• Arrays |
Electric Field |
|||
• Cellll Phone Circuitsi its |
||||
• Displaysi l |
Electric Field |
• Lab on Chipip |
|
|
|
Assisted |
|
|
|
|
Assisted Self-Assembly |
|
|
|
|
Self-Assembly |
|
|
|
|
“Pick & Place” |
|
|
|
|
"Pick & Place" |
|
|
|
|
Heterogeneous |
|
|
|
|
Heterogeneous |
Nano/Micro Scale |
||
True Molecularl l Scalele |
Integration |
|||
IntegrationProcessProcess |
Heterogeneous |
|||
Heterogeneoust |
||||
|
Integration |
|||
Integrationt ti |
|
|||
|
• DNA Optical Memory |
|||
• Molecularl l Electronicsl t i |
|
|||
|
Nanometert Scalele |
•Logic Devices |
|
|
|
|
|
||
|
Heterogeneoust |
|
|
|
|
Integrationt ti |
|
|
|
|
• Nanoelectronicsl t i |
|
|
|
|
• Nanotubet Devicesi |
|
|
|
FIGURE 6.3. Chart shows some of the potential advantages and applications for using electric field assisted self-assembly to carry out the heterogeneous integration of molecular, nanoscale and microscale components into higher order materials and devices. Electric field assisted heterogeneous integration technology has the hierarchical logic of allowing one to control the organization and communication of structures and components from molecular level —> nanoscale level —> “Highly Integrated 3-D Structures and Devices” <— micron scale components and lift-off type devices.
6.2. ACTIVE MICROELECTRONIC ARRAY HYBRIDIZATION TECHNOLOGY
A variety of microelectronic arrays have been designed and fabricated by Nanogen, primarily for DNA genotyping diagnostic applications (8–17). These includes an early stage 25 test site microelectronic array device, and more advanced arrays with 100, 400 and 10,000 test sites or microlocations (Figure 6.4). The 100 test-site chip, which has
FIGURE 6.4. Shows Nanogen’s 100 test site microelectronic array, a silicon wafer of 400 test site microarrays and a packaged 10,000 test site microarray. The 400 and 10,000 test site arrays have CMOS electronic control elements incorporated into the chips underlying structure.