

Computational Methods for Protein Structure Prediction & Modeling V1 - Xu Xu and Liang
.pdf5. Protein Structure Comparison and Classification |
157 |
5.3Multiple Structure Comparison and Structural Motif Search
Protein motifs, specifically active sites and binding sites, play important roles in biochemical reactions. Motifs are defined as substructures that are common to a set of proteins that possess functional or evolutionary relationships. Querying proteins for specific motifs and discovering motifs in a set of proteins are crucial for the functional classification and understanding of proteins. Since active sites of proteins are determined by structure of the participating amino acids rather than their sequence order, structural motifs can be more useful than sequence motifs. However, structural motif detection is more challenging. First, the size of the search space is much larger than in the sequence domain. Each residue can be one of 20 amino acids, so a motif of length 5 amounts to 205 possibilities in the sequence domain. This number is much larger for structural motifs if one considers the number of structural conformations of 5 residues without the sequence constraint. Also, for sequences, the type of each residue is known with almost certainty, but for structures the data has limited resolution. So even if two proteins have the same structural motif, their alignment may not produce the perfect score.
Multiple structure alignment of a set of related proteins results in a consensus structure which has the minimum RMSD sum to the protein structures in the set. [This is similar to finding the Steiner string for a set of sequences (Gusfield, 1997).]
5.3.1 Motif Detection
An important characteristic of motif finding programs is the granularity of the discovered motifs as shown in Fig. 5.6. Some of these algorithms consider only C atoms, while others consider all the atoms in the protein structures (Pennec and
Fig. 5.6 Motif definition for TESS program: relative conformation of the catalytic triads from chymotrypsin 1cho.
158 Orhan C¸ amo˘glu and Ambuj K. Singh
Ayache, 1998; Singh and Saha, 2003). Another set of methods defines motifs not by atoms but by secondary structure elements (Kato and Takahashi, 2001). Methods have also been proposed for the detection of small active sites while preserving atom types in motifs (Wallace et al., 1997). There are also methods that use both sequence and structure information simultaneously (Bradley et al., 2002). A majority of these methods adopt RMSD as an indication of quality.
Singh and Saha (2003) proposed a framework for structural motif queries. They incorporate the label information as well as the coordinates of the atoms. Starting from an initial alignment, the nearest neighbors of points in two structures are found and this information is used to modify the alignment. This process is carried out until convergence. The distance function on the structures is modified to account for the label information.
TESS (Wallace et al., 1997) is based on geometric hashing and finds small (usually two or three residues long) active sites. The method considers all of the atoms in the protein, and defines reference frames for each residue by using a combination of C, O, N, S atoms, depending on the residue types. For each reference frame, the relative positions of the atoms that are in the 18-A˚ neighborhood are stored in the grid. The grid positions that are heavily populated are analyzed. The types of atoms as well as their grid locations have to match for the resulting motif.
TRILOGY (Bradley et al., 2002) finds sequence–structure patterns, which are as small as three residues, across diverse families. In this automated approach, threeresidue patterns are extracted and a sequence feature of residue types and a structure feature of relative positions of the residues are computed for each pattern. Structure feature is based on the C –C distances between residues and, C –C vectors for each residue in the pattern. Sequence feature is based on the types of residues in the pattern and their distance from each other on the protein sequence. Residues are grouped into seven classes and these class types are used in the definition of sequence features to increase the flexibility of the representation. Patterns that have similar structure and sequence features are identified. These three-residue patterns are then extended by merging patterns that vary by a single residue. This process is carried out until the patterns fail to cover at least three SCOP superfamilies. At the final step, significance scores are assigned to these patterns based on the likelihood
of obtaining them at random. Figure 5.7 depicts a pattern whose sequence feature is Ax4−5[FY W ]x7−8 N .
Chen and Bahar (2004) proposed an unsupervised approach to discover frequent patterns in protein families. In this approach, each residue is characterized by its dynamic features, and biochemical and geometric features of the neighboring residues. Dynamic features summarize the rigidness of the local structure around the residue based on the interactions between neighboring residues; biochemical features summarize the amino acid types and chemical properties of neighboring residues; and geometric features summarize the relative positions of the neighboring residues in a similar manner to Pennec and Ayache (1998). After the feature extraction step, the frequency for each feature is computed. Features that occur at a frequency higher than a predefined threshold are considered to be frequent patterns. Then, these frequent patterns are merged to obtain longer augmented frequent patterns.

5. Protein Structure Comparison and Classification |
159 |
Fig. 5.7 Features of three-residue patterns used in TRILOGY program. Points represent C atoms and arrows represent C − C vectors (Bradley et al., 2002).
Jonassen et al. (1999) find local packing motifs in protein structures. For each residue, residues in close proximity are identified and an ordered list is created based on these residue types. Residues that have similar ordered lists are considered as candidates that participate in a local packing motif. The similarity of these candidates is finally verified in the structure domain by superimposing local structures around them.
5.3.2 Multiple Structure Alignment
Although many algorithms have been proposed for the pairwise structure alignment problem, there are only a few algorithms available for multiple structure alignment. A popular technique is to compute pairwise alignments, and to construct a multiple alignment from these (Orengo and Taylor, 1996; Gerstein and Levitt, 1996; Guda et al., 2001). Star alignment-based approaches are usually adopted for this construction. First, all pairwise comparisons are performed and a pivot protein, whose RMSD sum to other proteins is minimum, is chosen. Then, the proteins are aligned to the pivot and a multiple structure alignment is constructed. Figure 5.8 depicts the multiple alignment of 15 proteins to protein 1arb. A shortcoming of this approach is that it may miss global patterns since only two proteins are considered at a time. This is reminiscent of the problems arising during multiple alignment of sequences (Gusfield, 1997). A set of recent algorithms capture the global relationship by first extracting common substructures in protein structures and then by constructing multiple structure alignment using the common substructures (Leibowitz et al., 2001; Shatsky et al., 2002; Dror et al., 2003).
Gerstein and Levitt (1996) extend iterative dynamic programming to obtain a multiple structure alignment. Upon performing all pairwise alignments, the pivot that has the smallest average distance to other structures is found. Then, all structures are aligned to this structure. So, if position i in the median structure is aligned to position
jin the first structure, and to position k in the second structure, then positions j and
kare aligned to each other.

160 |
Orhan C¸ amo˘glu and Ambuj K. Singh |
Fig. 5.8 Multiple structure alignment of 10 proteins using 1arb as the pivot by using VAST. Molecular representation is made by Cn3D (http://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml).
Guda et al. (2001) extend CE (Shindyalov and Bourne, 1998) to perform multiple structure alignment. They first compute pairwise alignments of proteins, and choose a pivot structure. After an initial alignment is found using the pivot structure, it is refined by a Monte Carlo algorithm.
Leibowitz et al. (2001) use geometric hashing to find common substructures. These common substructures are used as a seed for the multiple alignment. The seeds are merged with each other to obtain larger seeds and the seed that produces the highest scoring alignment is reported. Shatsky et al. (2002) developed a technique that is faster and does not require all proteins to participate in the alignment. Each protein is used as pivot in turn and the one with the best multiple alignment is chosen. Dror et al. (2003) further improved these algorithms by computing the common core of the query proteins using secondary structure elements. The performance is better since SSEs are used instead of residues. In this approach, each SSE is represented as a line segment. A fingerprint for each SSE pair is computed based on the distance and angle between SSEs. These fingerprints are mapped to a 5D grid. Pairs in the same and adjacent buckets in this grid are used as the bases for multiple alignments. For each base, rigid body transformations of proteins are computed. The bases which have similar rigid body transformations of proteins are merged to obtain larger bases. These large bases are used as the common core of the multiple alignment.
5.4 Structure Search in Protein Databases
As the sizes of experimentally determined (Berman et al., 2000) and theoretically estimated (Pieper et al., 1999) protein structures grow, there is a need for scalable
5. Protein Structure Comparison and Classification |
161 |
searching techniques. Current pairwise alignment techniques produce high-quality pairwise alignments. However, such tools incur excessive running times for database queries, i.e., when a query protein is searched against a large target set. Current tools alleviate this problem by building online databases that contain precomputed results for known protein structures [MMDB (Wang et al., 2002) for VAST, FSSP (Holm and Sander, 1996) for DALI, web database for CE]. In general, scalable techniques are needed for:
1.Searching a protein against a large set of proteins,
2.Clustering a large set of protein structures,
3.Comparing two sets of proteins, and
4.Multiple structure alignment based on pairwise comparisons.
To solve the above problems, some index-based approaches have been proposed (Aung and Tan, 2004; Camoglu et al., 2004). ProtDex2 (Aung and Tan, 2004) uses an inverted file index to index features based on SSEs. It extracts features on SSEs by using their relative distances. The overall similarity of proteins is estimated and the promising candidates are compared using pairwise alignment techniques. Next, the approach developed in Camoglu et al. (2004) is discussed in detail. First, we discuss principles of index-based approaches, then we present an index-based structure comparison technique. The query algorithm is explained in Section 5.4.3, and experimental results are presented in Section 5.4.4.
5.4.1 Index Based Approaches
In numerous applications from databases to image analysis, index-based solutions have been applied to demanding search problems. The main advantage of using index structures is that they reduce the computational complexity of searching. A sequential search compares all of the n elements in the database with the query and reports the best results with a complexity of O(n). This complexity can be reduced to O(log(n)) using an index structure.
The goal of using an index structure is to reduce the number of pairwise comparisons by quickly selecting the promising candidates. In order to build an index structure, some features are extracted from proteins. Then, an index structure is built on the extracted features. Given a query protein, its features are extracted and the database index is queried with these features. Proteins whose features are similar to the query protein’s features are aligned using a more extensive algorithm, such as the ones described in Section 5.5.2.
5.4.2 PSI: An Index-Based Structure Comparison Program
PSI (Protein Structure Index) (Camoglu et al., 2004) is an SSE-based approach. It finds high-quality seeds by aligning the SSEs of a database protein with those of a given query protein. For a query protein (or a set of query proteins), this technique can be used to find similar proteins in a target dataset efficiently.
162 |
Orhan C¸ amo˘glu and Ambuj K. Singh |
PSI extracts feature vectors corresponding to triplets of neighboring SSEs. These triplets are regarded as the building blocks of the larger protein alignment. Later, these features are embedded in Euclidean space and stored in an index structure.
The index construction consists of the following four steps:
1.SSE approximation: The structural features of SSEs are simplified by representing them as line segments in 3D. This is achieved by first splitting up an SSE in two halves at the middle residue. Centers of mass are computed for each half, and a line segment is defined between them. This line segment is extended in both directions to cover the length of the SSE.
2.Triplet construction: Triplets of SSEs are used as primitive elements for building an alignment. A set of matching triplets between two protein structures can be used to align the given proteins using rigid body transformations (Arun et al., 1987). Since residues that are spatially close are more valuable for structure similarity
(Eidhammer and Jonassen, 2001), neighborhood constraints are imposed while building the SSE triplets. A sphere of radius 50 A˚ is centered at each SSE and up to four nearest SSEs are chosen. All possible pairs of these SSEs are combined with the center SSE to define the relevant triplets. An example of triplet construction is shown in Fig. 5.9. Empirically, the average number of triplets per SSE turns out to be 3.8 for the proteins in PDB.
3.Feature vector extraction: After the triplets of protein structures are determined, feature vectors are constructed. Let <si , s j , sk > be a triplet. The line segment approximation of each SSE is split into three equisized, non-overlapping intervals. The middle interval of each SSE is used to represent the SSE. The pair of SSEs, <si , s j >, contributes three values to the feature vector:
(a)mini j = minimum distance between the midregions of si and s j .
(b)maxi j = maximum distance between the midregions of si and s j .
(c)i j = the angle between the line segment approximations of si and s j .
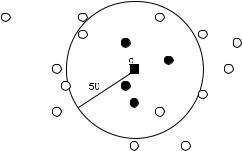
Fig. 5.9 The local neighborhood set of the SSE, c, of a protein. The black square corresponds to the midpoint of c. The circles represent the midpoints of the remaining SSEs of the same protein. 50 A˚ is the threshold distance for local neighborhood. Four best neighboring SSEs (shown with filled circles) are chosen.

5. Protein Structure Comparison and Classification |
163 |
Fig. 5.10 SSE pair <si , s j > contributes three values to the feature vector for triplet <si , s j , sk >: min(i, j) and max(i, j), minimum and maximum distance between the mid-portions of si and s j , and (i, j), angle between si and s j .
Figure 5.10 shows the extraction of these values for a pair of SSEs. This set of values is extracted for each pair in the triplet, resulting in a feature vector of size nine for each triplet.
4.Multidimensional index structure construction: The feature vector for an SSE triplet has three range values (minimum and maximum distance values) and three angle values. Each feature vector defines an extent in a six-dimensional Euclidean space. The feature vectors are embedded and indexed using an R*-tree (Beckmann et al., 1990). (R*-trees are dynamic index structures that provide efficient range search queries for multidimensional data.)
5.4.3 Query Algorithm
For a given query protein, the search technique (Camoglu et al., 2003) runs in four steps:
Step 1: Similar triplets between database proteins and query protein are computed and stored.
Step 2: A triplet pair graph (TPG) is constructed on similar triplet pairs. An example of such a graph is shown in Fig. 5.11.
Step 3: A bipartite graph is constructed using the TPG. The largest matching in this graph defines the initial alignment seed at SSE level. A p-value is computed for each seed. An example bipartite graph is shown in Figure 5.12.
Step 4: The proteins that have large p-values are removed without further consideration. The C alignment of the remaining proteins are determined using a pairwise structural alignment algorithm.

164 |
Orhan C¸ amo˘glu and Ambuj K. Singh |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 5.11 The triplet pairs between the two proteins and their scores are shown on the left. qi is a secondary structure element of query protein and pi is a secondary structure of target protein. The corresponding triplet pair graph (TPG) is shown on the right. The TPG has three connected components: one with four triplet pairs, the others with two and one.
Fig. 5.12 The bipartite graph of the largest weight component in Fig. 5.11 (assuming s1 + s2 + s3 + s4 + s7 > max(s6, s4 + s5)). There is a conflict in the alignment of SSE q1: it has an edge to both p1 and p11. The edge with the largest weight is chosen to resolve the conflict. Assuming s1 + s2 > s7, the initial alignment will be {q1 − p1, q2 − p2, q3 − p3, q4 − p4, q5 − p5}.
5.4.4Experimental Evaluation of PSI
The results of PSI are verified with SCOP classification and pairwise comparison tools. The first set of experiments analyzed how well the SCOP classification of a query protein matched with the SCOP classifications of the top-ranking proteins returned by PSI. On the average, 2.5 of the top 3 proteins and 5.4 of the top 10 proteins share the same superfamily as the query protein. This shows that PSI captures strong structure similarities. The same experiment was carried out for fold level classifications and similar results were obtained, showing the ability of PSI to capture remote homologies. Detailed explanation of these experiments can be found in Camoglu et al. (2004).
PSI was also compared with the pairwise comparison tools VAST, CE, and DALI. More than 98% of its results concurred with those of VAST. PSI ran 3 to 3.5

5. Protein Structure Comparison and Classification |
165 |
Fig. 5.13 The number of similar proteins of high significance for query protein 1d3g-A found by VAST, DALI, and CE, and the number of such proteins returned by PSI. For example, there are five proteins DALI and Vast found as similar but CE found dissimilar; all five of these are returned by PSI. Note that PSI returned all proteins for which VAST, DALI, and CE agree.
times faster than VAST’s pruning step. Similar results were also obtained for CE and DALI.
PSI can be incorporated with other tools to accelerate them by discarding dissimilar proteins. PSI-DALI (for DALI) and PSI-CE (for CE) were implemented in order to demonstrate this. PSI-DALI ran 2 times faster than DALI with 98% recall. PSI-CE ran 2.7 times faster than CE with 83–92% recall.
PSI has a different recall when compared to the results of VAST, CE, and DALI. An in-depth analysis revealed that these three comparison tools produce different results themselves. In Fig. 5.13, the results of these tools for query protein 1d3g-A, dihydroorotate dehydrogenase from human, are displayed. As can be seen, all tools agree over a large portion (75 proteins) of the result; PSI returns these proteins as well. On the whole, PSI is able to find the consensus set of results for each query.
5.5 Protein Classification
Although the study of a single structure or the alignment of a small group of structures can reveal a great deal of information, a global comprehensive view of the protein space is essential to understanding the fold similarities and the evolutionary process. Such a hierarchical classification of proteins based on the analysis of structure has been pursued by a number of researchers (Murzin et al., 1995; Orengo et al., 1997; Holm and Sander, 1996). The resulting organization of protein structures brings a semblance of order to a dynamic field and also provides a valuable resource to other biologists for benchmarking studies. In one such study, Chothia et al. (2003) study the evolution of protein domains across pathways and species. They also study the
166 |
Orhan C¸ amo˘glu and Ambuj K. Singh |
distribution of domain combinations. They infer a number of relationships such as power law behavior and preferential order of domain combinations based on the study. Gerstein (1997) analyzed representative proteomes from the three kingdoms of life for the SSE content, small motifs of SSEs (alpha-alpha-alpha or beta-alpha- beta), and folds. Though the genomes had similar SSE content, there were marked differences in the observed motifs.
In this section, we introduce various structure classification databases and an automated classification scheme for protein structures. Then, we describe the use of pairwise comparison tools as component classifiers. In Section 5.5.3, we discuss the use of decision trees in automated classification. Some experimental results are presented in Section 5.5.4.
5.5.1Structure Classification Databases
Protein structure classification databases employ different heuristics and similarity metrics, and can be fully automated (Holm and Sander, 1996), semiautomated (Orengo et al., 1997), or manual (Murzin et al., 1995).
SCOP (Structural Classification Of Proteins) (Murzin et al., 1995) is one of the most popular classification databases. It hierarchically classifies proteins into four categories:
Class: This is the highest level of the hierarchy. The main similarity criteria at this level are the type and the general layout of SSEs in the proteins. There are four main classes: (1) all , all the SSEs are helices, (2) all , all the SSEs are strands,
(3) / , helices and strands are in close proximity, (4) + , helices and strands are apart. In addition to these main classes, there are some other less populated classes (membrane, multiple domain, small protein, etc.).
Fold: This is the second level of the SCOP hierarchy. Members of the same fold have similar SSE arrangements (however, this similarity is not clearly defined). Disagreements between automated methods and SCOP appear mostly on this level. In general, fold-level relationships are observed between proteins that have possible remote evolutionary relationships.
Superfamily: Proteins in the same superfamily possess functional and evolutionary relationships. They have the same active sites, or participate in similar reactions and pathways. They have significant structural similarity and some sequence similarity. This level is one of the most interesting classification levels, because functional relations between proteins may be inferred using similarities at this level.
Family: This is the lowest level of classification. Members of a family possess strong evolutionary and functional relationships. Identification of family members is a fairly simple procedure since the main measure at this level is the sequence similarity. Proteins that have more than 30% sequence identity are classified into the same family. Proteins that have lower sequence similarity, but very strong structural similarity and functional relationships are also classified into the same family.