

Homogeneous Catalysis
.pdfWATER-GAS SHIFT REACTION AND RHODIUM-CATALYZED CARBONYLATION |
63 |
Figure 4.5 Proposed catalytic cycle for the water-gas shift reaction in the Monsanto process intermediates such as 4.5, 4.28, etc. may also be involved, but there is no direct evidence.
complex in the absence of CH3I. In a situation where no CH3I is available, the acetic acid–forming catalytic cycle (Fig. 4.1) ceases to exist. However, since the water-gas shift cycle continues to be operational, rhodium remains in solution and does not precipitate out.
At a molecular level, the intimate mechanism for the water-gas shift reaction with 4.1 as the precatalyst is not known in any detail. In the conversion of 4.1 to 4.12, the evolution of hydrogen is probably indicative of the intermediacy of a hydrido complex. Similarly, in the conversion of 4.13 to 4.1, where HI and CO2 are eliminated, nucleophilic attack on coordinated CO may be involved. Although these mechanistic conjectures are plausible, there is no direct evidence to support them.
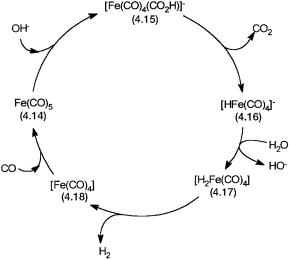
The water-gas shift reaction has also been studied under high pH with metal carbonyls as catalysts. The catalytic cycle with Fe(CO)5 as the precatalyst is shown in Fig. 4.6. This reaction with low turnover is carried out at 130–180 C, under 10–40 bar of CO, with alkali metal hydroxide as a promoter.
In situ IR spectroscopy shows the presence of 4.14 and 4.16 in the catalytic mixture. The presence of 4.15 is inferred from the known chemistry of metal carbonyls. Both nucleophilic attacks on coordinated CO by HO and decarboxylation of the resultant complex as shown in 4.8 are well-precedented reactions.
64 CARBONYLATION
Figure 4.6 Proposed catalytic cycle for the water-gas shift reaction by Fe(CO)5 at high pH.
4.4 FISCHER–TROPSCH REACTION AND COBALTCATALYZED CARBONYLATION
The Fischer–Tropsch reaction, named after the original inventors, is essentially the conversion of synthesis gas (CO H2) to a mixture of hydrocarbons, and to a lesser extent oxygenated hydrocarbons. The commercial Fischer–Tropsch reaction such as the one practiced at SASOL in South Africa uses potassiumand copper-promoted heterogeneous iron catalysts. A wide range of hydrocarbons, containing one to more than 100 carbon atoms, are produced. Paraffins and to a lesser extent alkenes are the main products. These are thought to arise through carbide intermediates that are converted to CH2 groups. There is no homogeneous Fischer–Tropsch catalyst that gives paraffin or alkene in good yield.
The CO used for the carbonylation reaction always contains some hydrogen. The side products in the BASF carbonylation process arise due to Fischer– Tropsch reaction catalyzed by the cobalt catalyst. The high temperatures and pressures used in the BASF process are conditions under which the Fischer– Tropsch reaction with soluble cobalt catalyst can take place. In the Monsanto process the reaction conditions are much milder, and the side-product-forming Fischer–Tropsch reaction is avoided.
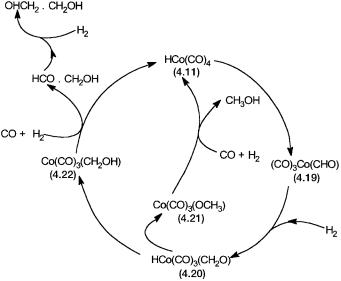
Based on the known reactions of cobalt carbonyls, the catalytic cycle shown in Fig. 4.7 has been suggested. The hydrido carbonyl 4.11 is converted to 4.19, a formyl species. Formyl complexes are considered to play a key role in all
FISCHER–TROPSCH REACTION AND COBALT-CATALYZED CARBONYLATION |
65 |
Figure 4.7 Proposed catalytic cycle for the Fischer–Tropsch reaction, giving oxygenated hydrocarbons.
homogeneous catalytic reactions that give mainly oxygenated hydrocarbons from synthesis gas. Intramolecular CO insertion into a metal–hydride bond is thermodynamically unfavorable under mild conditions (see Section 2.3.2). However, under high pressure and temperature this reaction may be kinetically favorable or may take place by an intermolecular mechanism. The formyl complex reacts with dihydrogen to give 4.20, a complex with coordinated formaldehyde. Two different reactions leading to the formation of 4.21 or 4.22 are possible. The hydroxymethyl complex 4.22 is responsible for the formation of ethylene glycol through the intermediate formation of glycol aldehyde. On the other hand, 4.21 produces methanol.
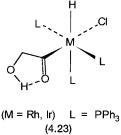
To this date most of the reactions, though reasonable from the point of view of established organometallic chemistry of cobalt and other metals, remain speculative. Formyl, formaldehyde, hydroxymethyl, and methoxy complexes of transition metals are known, and this to a large extent gives confidence about the correctness of the proposed catalytic cycle. For example, the production of ethylene glycol from 4.22 must proceed through a catalytic intermediate, where the carbon atom of the hydroxymethyl group forms a C–C bond with a carbonyl or a formyl group. Such complexes are indeed known and fully characterized for rhodium and iridium. As shown for 4.23, part of the stability of such complexes may be due to intramolecular hydrogen bonding.
66 CARBONYLATION
In the 1970s Union Carbide had reported the use of rhodium with promoters such as amines, carboxylates, etc. for the synthesis of ethylene glycol from CO plus H2. Manufacture of ethylene glycol by this route, however, was never commercialized. The mechanism of this reaction is not understood. Both mononuclear and polynuclear (cluster) rhodium carbonyls can be seen by NMR and IR spectroscopy under conditions approximating that of the catalytic reaction. The question as to whether the catalytic intermediates are mononuclear or cluster has not been answered with any certainty so far.
Some mechanistic information is available on ruthenium-based homogeneous Fischer–Tropsch reactions. By in situ IR spectroscopy, in the absence of any promoter, only Ru(CO)5 is observed. An important difference between the cobalt and the rhodium system on the one hand and ruthenium on the other is that in the latter case no ethylene glycol or higher alcohols are obtained. In other words, in the catalytic cycle the hydroxymethyl route is avoided.
With the ruthenium-based catalyst, in the presence of a promoter such as I both activity and selectivity towards oxygenated two-carbon-containing products increase. Under these conditions in situ IR spectroscopy shows the presence of the polynuclear complex [HRu3(CO)11] and the mononuclear [Ru(CO)3I3] . These complexes themselves are not catalytically active intermediates; rather they are the precursors of such intermediates. From independent experiments it appears that [HRu(CO)4] and [Ru(CO)4I2] are two of the actual active intermediates that are formed from the cluster and the mononuclear complexes, respectively. These complexes are not detected by spectroscopy, presumably because of insufficient concentration. There is also evidence to suggest that [HRu(CO)4] and [Ru(CO)4I2] react to give a formyl species. In other words, the first proposed step for the observed selectivity is an intermolecular reaction between [HRu(CO)4] and Ru(CO)4I2. Once the formyl species is formed, it is thought to undergo reactions similar to the ones shown in Fig. 4.7 to give oxygenated hydrocarbons.
4.5 RHODIUM-CATALYZED CARBONYLATION OF OTHER ALCOHOLS
Rhodium-catalyzed carbonylation of alcohols such as ethanol, n-propanol, and i-propanol have been studied. Unlike methanol carbonylation, central to all

FISCHER–TROPSCH REACTION AND COBALT-CATALYZED CARBONYLATION |
67 |
Figure 4.8 Hydrocarboxylation and carbonylation of propylene and n-propyl iodide. The outer cycle is the hydrocarboxylation pathway, while the inner cycle is the carbonylation pathway. The carbonylation pathway generates only n-butyric acid iodide. Note that 4.27 is identical with 4.5. A different designation is used to avoid crossreferencing.
these reactions is the reaction of alkenes with hydrido complexes that act as catalytic intermediates. Reactions where a carboxylic acid is formed from the reaction of an alkene with water and CO, as in reaction 4.9, is known as hydrocarboxylation (see Section 1.5). The outer catalytic cycle in Fig. 4.8
shows the hydrocarboxylation route of propylene to n-butyric and i-butyric acid iodides. The inner cycle is the carbonylation route of n-propyl iodide to n- butyric acid iodide.
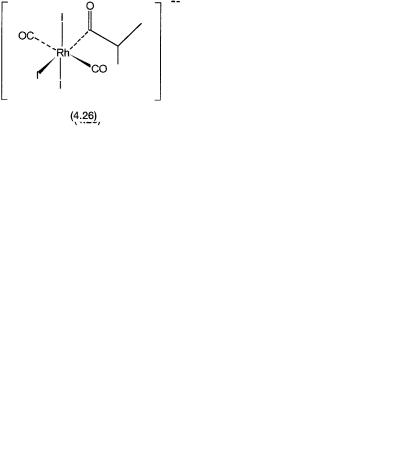
Although at a first glance the cycles may appear to be too complex, most of the catalytic species are analogues of intermediates shown in Fig. 4.2. Complexes 4.24 and 4.27 are oxidative addition products of 4.1 with n-propyl iodide and HI, respectively.
68 CARBONYLATION
The latter complex undergoes CO loss to generate coordinatively unsaturated 4.28. Conversion of 4.28 to 4.30 is the crucial step that is responsible for the formation of the branched isomer. Obviously this reaction is possible only when propylene is present as one of the reactants, or under reaction conditions where propylene from n-propanol is generated in situ. Conversion of 4.28 to 4.30 is an example of alkene insertion into an M–H bond in a Markovnikov manner (see Section 5.2.2 for a discussion on Markovnikov and anti-Markovnikov insertion). The anti-Markovnikov path leads to the formation of 4.29, which is in equilibrium with 4.24. Complexes 4.25 and 4.26 are analogues of 4.4 with n-butyl and i-butyl groups in the place of methyl. They reductively eliminate the linear and branched acid iodides. In the presence of water the acid iodides are hydrolyzed to give n-butyric and i-butyric acids.
4.6CARBONYLATION OF METHYL ACETATE
Conventional manufacture of acetic anhydride is based on the reaction of ketene (H2C—C—O) with acetic acid. Eastman Chemical Company, a division of Eastman Kodak, set up facilities to manufacture 500 million lbs/yr of acetic anhydride and 165 million lbs/yr of acetic acid using coal as the feedstock in 1984. In this process coal is gasified to give synthesis gas, which is then converted to methanol by a heterogeneous catalytic process. Reaction of methanol with acetic acid gives methyl acetate, which is carbonylated to give acetic anhydride. Eastman Kodak consumes more than 1 billion lbs/yr of acetic anhydride in the production of cellulose esters used in the production of photographic films, plastics, coating chemicals, etc. The major factors behind the spectacular success of this process are as follows. All the production facilities are located in the heart of Appalachian coalfields. This process is less energy
CARBONYLATION OF METHYL ACETATE |
69 |
intensive than the ketene-based process. The carbonylation process recycles acetic acid, a coproduct in the manufacture of cellulose esters, in the synthesis of methyl acetate. Finally, conversions and rhodium metal recovery efficiencies of this process are sufficiently high to make the overall economics viable and attractive.
4.6.1Mechanism and Catalytic Cycle
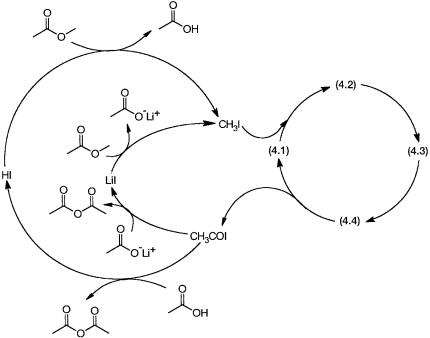
The acetic acid–forming part of the catalytic cycle for methanol carbonylation consists of reactions between acetyl iodide and water to give acetic acid and HI (Fig. 4.2, bottom left). The hydroiodic acid reacts with methanol to regenerate CH3I and water. A similar mechanism operates for the carbonylation of methyl acetate. Acetic acid and acetyl iodide react to give acetic anhydride and HI. The latter reacts with methyl acetate to regenerate acetic acid and methyl iodide. These reactions are shown in Fig. 4.9 by the large, left-hand-side loop.
Figure 4.9 Carbonylation of methyl acetate to acetic anhydride. The right-hand-side cycle is the same as in 4.1 and 4.2. The big cycle on the left is similar to the organic cycle of 4.2, where acetic acid and methyl acetate substitute for water and methanol. The small inner cycle on the left is the dominant product-forming pathway.
70 CARBONYLATION
In addition to this, there is another lithium salt promoted pathway (Fig. 4.9) that contributes significantly to product formation. Here the product-forming reaction between lithium acetate and acetyl iodide is followed by the reaction between LiI and methyl acetate. These reactions are shown by the inner loop on the left-hand side. In fact, the inner loop is the dominant product-forming pathway, and lithium salts play a crucial role in the overall catalysis. Note that the right-hand-side loop of the catalytic cycle is exactly the same as in Fig. 4.1(a).
Evidences for the proposed catalytic cycles come from kinetic and spectroscopic studies. In situ IR investigation indicates the presence of 4.1 as the main catalytic intermediate. The rate of acetic anhydride formation shows a complex dependence on the concentrations of rhodium, CH3I, and lithium. Under conditions where lithium concentration is high, the rate is first order with respect to rhodium and CH3I; that is, the rate is given by Eq. 4.3. However, with low lithium concentration the rate is independent (i.e., zero order) with respect to rhodium and methyl iodide concentrations. These observations are easily explained by identifying the slowest step under these two sets of conditions. With enough lithium the rates of reactions involving lithium salts are obviously high. Under these conditions oxidative addition of CH3I to 4.1 is the slowest step, and rate expression 4.3 is obeyed. With low lithium concentration any one of the steps that involve lithium salts becomes the rate-determining one. The overall rate in such a situation is independent of rhodium and CH3I concentrations.
4.7 CARBONYLATION OF ALKYNES; MANUFACTURE OF METHYL METHACRYLATE
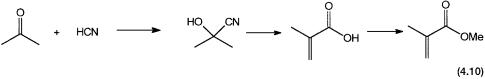
The polymer of methyl methacrylate (MMA) is known as “Perspex.” It is a clear transparent glasslike material with high hardness, resistance to fracture, and chemical stability. The conventional route, as shown by reaction 4.10, involves the reaction between acetone and hydrocyanic acid, followed by sequential hydrolysis, dehydration, and esterification. This process generates large quantities of solid wastes. An alternative route based on a homogeneous palladium catalyst has recently been developed by Shell. In this process a palladium complex catalyzes the reaction between propyne (methyl acetylene), methanol, and carbon monoxide. This is shown by reaction 4.11. The desired product is formed with a regioselectivity that could be as high as 99.95%.

CARBONYLATION OF ALKYNES; MANUFACTURE OF METHYL METHACRYLATE |
71 |
This invention has its roots in Reppe chemistry. In the late 1930s, Reppe in Germany had developed a number of manufacturing processes for bulk chemicals, where acetylene was used as one of the basic building blocks. Even today BASF and Rohm Hass manufacture large quantities of acrylic acid and its esters by hydrocarboxylation of acetylene. This reaction, 4.12, is catalyzed by a mixture of NiBr2 and CuI. It involves high pressure (100 bar) and temperature (220 C), and mechanistically is not fully understood.
In contrast, the Shell process for MMA operates under milder conditions (60 C, 10–60 bar), and the mechanism at a molecular level is better understood. The reaction is usually carried out in methanol, which acts both as a solvent and as a reactant. The precatalyst is Pd(OAc)2, which is mixed with an excess of phosphine ligand to generate the active catalytic intermediates in situ. An important requirement for efficient catalysis is the presence of an acid HX that acts as a co-catalyst.
4.7.1Mechanism and Catalytic Cycle
The Shell process uses a novel ligand, 4.31, a phosphine that also has a pyridine ring. This ligand in the presence of an acid may act as a bidentate as well as a monodentate ligand. This is because the acid protonates the nitrogen atom of the pyridine ring and an equilibrium, as shown by 4.13 is set up.
Under this condition both chelation, as shown by 4.32, and monodentate coordination, as shown by 4.33, may occur. Although not shown, in 4.33 the

72 CARBONYLATION
fourth coordination site is occupied by a solvent molecule or weakly coordinating X . A hypothetical catalytic cycle, based on these considerations, is shown in Fig. 4.10. In its protonated form ligand 4.31 acts as a labile, weakly coordinating ligand, easily displaced by reactants such as CO, methyl acetylene, etc.
As shown in the proposed cycle, mixing Pd(OAc)2 with excess ligand and a noncoordinating acid HX in methanol probably produces a species such as 4.34. Substitution of the protonated ligand by CO gives 4.35. Carbon monoxide insertion into the Pd–OMe bond and coordination by the protonated ligand produces 4.36. The ligand is displaced again by methyl acetylene to give 4.37. Insertion of the alkyne into the Pd–CO2Me bond, and coordination by the displaced ligand gives 4.38. A proton transfer from the protonated ligand to the -bonded alkene leads to the formation of MMA and 4.39. The latter reacts with methanol to complete the catalytic cycle and regenerate 4.34.
The catalytic cycle, though reasonable, is hypothetical. The proposed reactions and complexes have precedents in organometallic chemistry. The rate of the overall reaction is first order with respect to methyl acetylene and independent (zero order) of acid concentration as long as sufficient acid and 4.31 are present. Indirect but strong evidence for the proposed mechanism comes from structural modifications of the ligand and effects of such modifications