

Olah G.A. - A Life of Magic Chemistry (2001)(en)
.pdf70 A L I F E O F M A G I C C H E M I S T R Y
lish much of my work and to lecture at different international symposia and meetings, such as the IUPAC Congresses in Paris (1957), Munich (1959), and London (1963); the Cork (Ireland) Conference on Physical Organic Chemistry (1961); and various meetings and symposia of chemical societies. I also used these occasions to visit and give lectures at many universities. My personal contacts and my circle of colleagues, many of whom over the years became friends, grew.
In the summer of 1963, I learned that I had won the American Chemical Society Award in Petroleum Chemistry for my work on Friedel-Crafts chemistry. It was a most welcome recognition for someone who only a few years earlier had fled his native country and started all over on a far-away continent. Although I have received numerous other awards and recognitions over the years, with the exception of the Nobel Prize, no other award touched me as much. I remember that my first ACS award carried with it a check for $5,000. My research director for some reason believed that a company employee was not
1960 with Georg Wittig

M O V E T O N O R T H A M E R I C A 71
In my ‘‘sailing’’ outfit, complete with jacket and tie, with Phil Skell and Heini Zollinger. Cork, Ireland, July 1964
At London IUPAC congress 1963

72 A L I F E O F M A G I C C H E M I S T R Y
With my father in Austria during a European visit in 1965.
entitled to any external ‘‘income’’ and I should give this to the company. He also raised some questions concerning royalties of any books I was writing in my spare time. The matter was finally settled by the president of the company, who found that these questions had no merit. I was able to keep my award money and modest book royalties (which I believe in any case would not have made any impact on the bottom line of the company’s earnings).
Concerning my research during my Dow years, as I discuss in Chapter 4, my search for cationic carbon intermediates started back in Hungary, while I was studying Friedel-Crafts-type reactions with acyl and subsequently alkyl fluorides catalyzed by boron trifluoride. In the course of these studies I observed (and, in some cases, isolated) intermediate complexes of either donor-acceptor or ionic nature.
Many elements readily form ionic compounds such as table salt (Na Cl ), in which the cationic sodium and anionic chlorine are held

M O V E T O N O R T H A M E R I C A 73
together by electrostatic, ionic bonding. Carbon, however, was long considered to lack the ability to form similar ionic compounds, except in very specific, highly stabilized systems, such as triphenylmethyl dyes.
The Chicago chemist Stieglitz (whose noted photographer brother was the husband of the painter Georgia O’Keefe) suggested in 1899 the possibility of ionic carbon compounds, but it aroused no interest. When I gave the Stieglitz memorial lecture in the 1980s at the University of Chicago and reminded the audience of Stieglitz’s pioneering idea, there was also little appreciation of it. It was in 1901 that Norris and Kehrman, as well as Wentzel, independently discovered that colorless triphenylmethyl alcohol gave deep yellow solutions in concentrated sulfuric acid. Triphenylmethyl chloride similarly formed orange complexes with aluminum and tin chlorides.
Von Baeyer (Nobel Prize, 1905) should be credited for having recognized in 1902 the saltlike character of the compounds formed. He then suggested a correlation between the appearance of color and salt formation—the so-called ‘‘halochromy.’’ Gomberg (who had just shortly before discovered the related stable triphenylmethyl radical), as well as Walden, contributed to the evolving understanding of the structure of related cationic dyes such as malachite green.
Whereas the existence of ionic triarylmethyl and related dyes was thus established around the turn of the twentieth century, the more general significance of carbocations in chemistry long went unrecognized. Triarylmethyl cations were considered an isolated curiosity of chemistry, not unlike Gomberg’s triarylmethyl radicals. Not only were simple hydrocarbon cations believed to be unstable, even their fleeting existence was doubted.
One of the most original and significant ideas in organic chemistry was the suggestion by Hans Meerwein that carbocations (as we now call all the positive ions of carbon compounds) might be intermediates in the course of reactions that start from nonionic reactants and lead to nonionic covalent products.
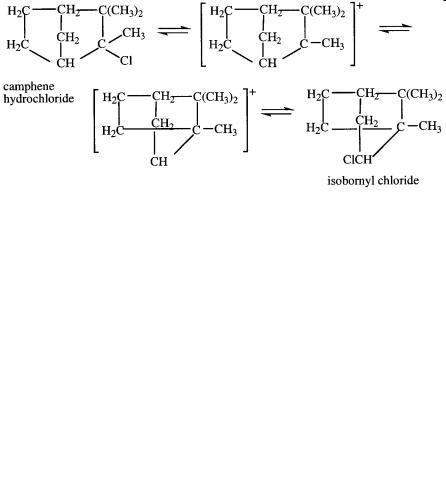
74 A L I F E O F M A G I C C H E M I S T R Y
In 1923, Meerwein, while studying the Wagner rearrangement of camphene hydrochloride to isobornyl chloride with van Emster, found that the rate of the reaction increased with the dielectric constant of the solvent. Furthermore, he found that certain Lewis acid chlorides such as SbCl5, SnCl4, FeCl3, AlCl3, and SbCl3 (but not BCl3 or SiCl4) as well as dry HCl, which promote the ionization of triphenylmethyl chloride by formation of carbocationic complexes, also considerably accelerated the rearrangement of camphene hydrochloride to isobornyl chloride. Meerwein concluded that the isomerization actually does not proceed by way of migration of the chlorine atom but by a rearrangement of a cationic intermediate. Hence, the modern concept of carbocationic intermediates was born. Meerwein’s views were, however, greeted with much skepticism by his peers in Germany, discouraging him from following up on these studies (see Chapter 9).
Ingold, Hughes, and their collaborators in England, starting in the late 1920s, carried out detailed kinetic and stereochemical investigations on what became known as nucleophilic substitution at saturated carbon and polar elimination reactions. Their work relating to unimolecular nucleophilic substitution and elimination, called SN1 and E1 reactions, in which formation of carbocations is the slow ratedetermining step, laid the foundation for the role of electron-deficient carbocationic intermediates in organic reactions.
Frank Whitmore in the United States in the 1930s in a series of papers, generalized these concepts to include many other organic reactions. Carbocations, however, were generally considered to be unstable

M O V E T O N O R T H A M E R I C A 75
and transient (short lived) because they could not be directly observed. Many leading chemists, including Roger Adams, determinedly doubted their existence as real intermediates and strongly opposed even mentioning them. Whitmore, consequently, was never able, in any of his papers in the prestigious Journal of the American Chemical Society, to use the notation of ionic R3C . The concept of carbocations, however, slowly grew to maturity through kinetic, stereochemical, and product studies of a wide variety of reactions. Leading investigators such as P. D. Bartlett, C. D. Nenitzescu, S. Winstein, D. J. Cram, M. J. S. Dewar, J. D. Roberts, P. v. R. Schleyer, and others contributed fundamentally to the development of modern carbocation chemistry. The role of carbocations as one of the basic concepts of modern chemistry has been well reviewed. With the advancement of mass spectrometry, the existence of gaseous carbocations was proven, but this could not give an indication of their structure or allow extrapolation to solution chemistry. Direct observation and study of stable, long-lived carbocations, such as alkyl cations in the condensed state, remained an elusive goal.
My work on long-lived (persistent) carbocations dates back to the late 1950s at Dow and resulted in the first direct observation of alkyl cations. Subsequently, a wide spectrum of carbocations as long-lived species was studied using antimony pentafluoride as an extremely strong Lewis acid and later using other highly acidic (superacidic) systems.
Until this time alkyl cations were considered only transient species. Their existence had been indirectly inferred from kinetic and stereochemical studies, but no reliable spectroscopic or other physical measurements of simple alkyl cations in solution or in the solid state were obtained.
It was not fully realized until my breakthrough using superacids (vide infra) that, to suppress the deprotonation of alkyl cations to olefins and the subsequent formation of complex mixtures by reactions of olefins with alkyl cations, such as alkylation, oligomerization, polymerization, and cyclization, acids much stronger than those known and used in the past were needed.

76 A L I F E O F M A G I C C H E M I S T R Y
Finding such acids (called ‘‘superacids’’) turned out to be the key to obtaining stable, long-lived alkyl cations and, in general, carbocations. If any deprotonation were still to take place, the formed alkyl cation (a strong Lewis acid) would immediately react with the formed olefin (a good -base), leading to the mentioned complex reactions.
In Friedel-Crafts chemistry it was known that when pivaloyl chloride is reacted with aromatics in the presence of aluminum chloride, tert- butylated products are obtained in addition to the expected ketones. These were assumed to be formed by decarbonylation of the intermediate pivaloyl complex or cation. In the late 1950s I returned to my earlier investigations of Friedel-Crafts complexes and extended them by using IR and NMR spectroscopy. I studied isolable complexes of acyl fluoride with Lewis acid fluorides, including higher-valence Lewis acid fluorides such as SbF5, AsF5, and PF5. In the course of these studies, it was not entirely unexpected that the generated (CH3)3CCOFSbF5 complex showed a substantial tendency toward decarbonylation. What was exciting, however, was that it was possible to follow this process by NMR spectroscopy and to observe what turned out to be the first stable, long-lived alkyl cation salt, namely, tert-butyl hexafluoroantimonate.
This breakthrough was first reported in 1962 and was followed by further studies that led to methods for preparing varied long-lived alkyl cations in solution.
The idea that ionization of alkyl fluorides to stable alkyl cations could be possible with an excess of strong Lewis acid fluoride that also serves as solvent first came to me in the early 1950s while I was still working in Hungary and studying the boron trifluoride-catalyzed alkylation of aromatics with alkyl fluorides. In the course of these studies I attempted to isolate RF : BF3 complexes. Realizing the difficulty of finding suitable solvents that would allow ionization but at the same would not react with developing, potentially highly reactive alkyl cations, I condensed alkyl fluorides with neat boron trifluoride at low temperatures. I had, however, no access to any modern spectrometers

M O V E T O N O R T H A M E R I C A 77
to study the complexes formed. I remember a visit at the time by Costin Nenitzescu (an outstanding but not always fully recognized Romanian chemist, who carried out much pioneering research on acid-catalyzed reactions). We commiserated on our lack of access to even an IR spectrometer. (Nenitzescu later recalled sending his cyclobutadiene-Ag complex on the Orient Express to a colleague in Vienna for IR studies, but the complex decomposed en route.) All I could do at the time with my RF-BF3 complexes was to measure their conductivity. The results showed that methyl fluoride and ethyl fluoride complexes gave low conductivity, whereas the isopropyl fluoride and tert-butyl fluoride complexes were highly conducting. The latter systems, however, also showed some polymerization (from deprotonation to the corresponding olefins). The conductivity data thus must have been to some degree affected by acid formation.
During a prolonged, comprehensive study some years later at the Dow laboratory of numerous other Lewis acid halides, I finally hit on antimony pentafluoride. It turned out to be an extremely strong Lewis acid and, for the first time, enabled the ionization of alkyl fluorides to stable, long-lived alkyl cations. Neat SbF5 solutions of alkyl fluorides are viscous, but diluted with liquid sulfur dioxide the solutions could be cooled and studied at 78 C. Subsequently, I also introduced even lower-nucleophilicity solvents such as SO2ClF or SO2F2, which allowed studies at even lower temperatures. Following up the observation of the decarbonylation of the pivaloyl cation that gave the first spectral evidence for the tertiary butyl cation, tert-butyl fluoride was ionized in excess antimony pentafluoride. The solution of the tert-butyl cation turned out to be remarkably stable, allowing chemical and spectroscopic studies alike.
In the late 1950s the research director of our laboratory was not yet convinced of the usefulness of NMR spectroscopy. Consequently, we had no such instrumentation of our own. Fortunately, the Dow laboratories in Midland just 100 miles away had excellent facilities run by E. B. Baker, a pioneer of NMR spectroscopy, who offered his help. To

78 A L I F E O F M A G I C C H E M I S T R Y
probe whether our SbF5 solution of alkyl fluorides indeed contained alkyl cations, we routinely drove in the early morning to Midland with our samples and watched Ned Baker obtain their NMR spectra. tert- Butyl fluoride itself showed a characteristic doublet in its 1H NMR spectrum due to the fluorine-hydrogen coupling ( JH.F = 20 Hz). In SbF5 solution, the doublet disappeared and the methyl protons became significantly deshielded from about 1.5 to 4.3. This was very encouraging but not conclusive proof of the presence of the tert-butyl cation. If one assumes that with SbF5 tert-butyl fluoride forms only a polarized donor-acceptor complex, which undergoes fast fluorine exchange (on the NMR time scale), the fluorine-hydrogen coupling would be ‘‘washed out,’’ while a significant deshielding of the methyl protons would still be expected. The differentiation of a rapidly exchanging polarized donor-acceptor complex from the long-sought-after ionic t-C4H9 SbF6 thus became a major challenge.
Ned Baker, himself a physicist, showed great interest in our chemical problem. To solve it, he devised a means to obtain the carbon-13 NMR spectra of our dilute solutions, an extremely difficult task at the time before the advent of Fourier transform NMR techniques. Labeling with carbon-13 was possible at the time only to about a 50% level (from Ba13CO3). When we prepared 50% 13C-labeled tert-butyl fluoride, we could, however, obtain at best only a 5% solution in SbF5. Thus the 13C content in the solution was tenfold diluted. However, Baker, undaunted, devised what became known as the INDOR (internuclear double resonance) method. Using the high sensitivity of the proton signal, he was able with the double-resonance technique to observe the 13C shifts of our dilute solutions—a remarkable achievement around 1960. The carbon-13 shift of the tertiary carbon atom in (CH3)3CFSbF5 of 13C 335.2 turned out to be more than 300 ppm deshielded from that of the covalent starting material. Such very large chemical deshielding (the most deshielded 13C signal at the time) could not be reconciled with a donor-acceptor complex. It indicated rehybridization

M O V E T O N O R T H A M E R I C A 79
from sp3 to sp2 and at the same time showed the effect of significant positive charge on the carbocationic carbon center.
Besides the tert-butyl cation, we also succeeded in preparing and studying the related isopropyl and the tert-amyl cations. The isopropyl cation was of particular relevance.
Whereas in the tert-butyl cation the methyl protons are attached to carbons that are only adjacent to the carbocationic center, in the isopropyl cation a proton is directly attached to the center. When we obtained the proton NMR spectrum of the i-C3H7F-SbF5 system, the CH proton showed up as an extremely deshielded septet at 13.0 ppm, ruling out a polarized donor-acceptor complex and indicating the formation of the (CH3)2CH ion. The 13C NMR spectrum was also conclusive, showing a very highly deshielded (by > 300 ) C atom ( 13C 320.6). The spectrum of the tert-amyl cation showed an additional interesting feature due to the strong long-range H-H coupling of the methyl protons adjacent to the carbocationic center with the methylene protons. If only the donor-acceptor complex were involved, such longrange coupling through an sp3 carbon would be small (1–2 Hz). Instead, the observed significant coupling ( JH-H = 10 Hz) indicated that the species studied indeed had an sp2 center through which the longrange H-H coupling became effective. Figure 6.1 reproduces the 1H NMR spectra of the tert-butyl, tert-amyl, and isopropyl cations. These original spectra are framed and hang in my office as a memento, as are the ESCA spectra of the tert-butyl and of the norbornyl cation (vide infra).
Our studies also included IR spectroscopic investigation of the observed ions (Fig. 6.2). John Evans, who was at the time a spectroscopist at the Midland Dow laboratories, offered his cooperation and was able to obtain and analyze the vibrational spectra of our alkyl cations. It is rewarding that, some 30 years later, FT-IR spectra obtained by Denis Sunko and his colleagues in Zagreb with low-temperature matrixdeposition techniques and Schleyer’s calculations of the spectra showed good agreement with our early work, considering that our work was