

Rauk Orbital Interaction Theory of Organic Chemistry
.pdfTRANSITION METAL BONDING TO ALKENES: ZEISE'S SALT |
189 |
fall near a ÿ 1:5jbj. The relative vertical spacing of the metal valence orbitals of the T- shaped …C2v† fragment is as shown in Figure 13.4. On this basis, the upper of the two a1 orbitals, n dz2 and dx2 ÿ y2, is placed near aC, the zero of the jbj scale. In PtClÿ3 , platinum is in the ‡2 formal oxidation state and contributes 8e, all of which are assigned to the valence levels. Therefore, the two a1 levels, n dz2 and dx2 ÿ y2, are the HOMO and LUMO, respectively. It is clear that the principal attractive interaction, shown by solid lines, is between the HOMO of ethylene and the LUMO of the metal. Optimum overlap between the p orbital and the dx2 ÿ y2 occurs if the ethylene is oriented perpendicular to the xy plane (the plane of the metal complex). This orientation also minimizes the repulsive interaction with the n dz2 orbital. It is the observed structure of Zeise's salt [281]. Back donation from the occupied b1 ˆ dxz orbital of the metal to the p of ethylene, shown by the dashed lines in Figure 13.7, is not very important because of the large
Ê
energy separation of the two orbitals. The CÐC distance of the bound ethylene, 1.37 A
Ê
[281], is similar to that in free ethylene, 1.34 A. The structure is best described as a p complex (rather than a metalacyclopropane). These conclusions are essentially the same as reached from DFT calculations [282], although greater signi®cance was attributed to the extent of p back bonding on the basis of B3LYP calculations [283].
We note two features of the bonding picture of Zeise's salt (and similar complexes). First, the LUMO of the complex is predominantly the p orbital of ethyleneÐit is lowered in energy by interaction with the pz orbital. One might expect reactivity toward nucleophiles. As we shall see in the next sections, intramolecular nucleophilic attack at the alkene carbon atom by hydride or alkyl ligands on the metal is a key step in ole®n polymerization. Second, the n dz2 orbital is raised in energy by interaction with the p orbital of ethylene to quite a high value and is the HOMOÐthe square planar Pt complex should be quite reactive toward Lewis acids from the direction perpendicular to the plane. In fact, the presence of the low-lying pz orbital complicates matters since attachment of an electrophile to one face of the square-planar complex lowers the symmetry and permits mixing of the two, resulting in a very much lower empty pd hybrid and attachment of a nucleophilic sixth ligand and a formal oxidation state of Pt(IV). The pentacoordinated species has to be regarded as a reactive intermediate [284]. The observed trans addition of HCl to square planar Pt(II) complexes is consistent with this expectation [285].
The perpendicular orientation of the alkene in such complexes is favored because it maximizes the overlap of the p bond with the LUMO (dx2 ÿ y2, Figure 13.7) and minimizes 4e repulsive interactions with the HOMO …n dz2†. The in-plane orientation is not expected to be strongly disfavored, however, because of the secondary interaction between the p orbital and the dxy orbital. The rotational barrier of ethylene in Zeise's anion was theoretically estimated to be 55 kJ/mol [282], within the range 42±63 kJ/mol measured by NMR for related complexes [286].
The binding of ethylene in Zeise's-like anions was investigated as a function of metal (Ni, Pd, Pt) and ligand (Clÿ, NH3) by B3LYP calculations [283]. The binding energy of ethylene to Zeise's anion, PtClÿ3 , was found to be 142 kJ/mol and was calculated to decrease in the series, PdClÿ3 (79 kJ/mol) and NiClÿ3 (26 kJ/mol). Substitution of chloride for ammonia, a poorer s donor, led to increases in binding energies but did not change the relative order of stabilitiesÐbinding energies of ethylene to c-MCl(NH3)‡2 : Pt, 193; Pd, 142; Ni 105 [283]. We shall attempt to rationalize these trends on the basis of orbital interaction theory.
The e¨ect of variation of metal and ligand on the energies of the orbitals of the T- shaped complex is shown in Figure 13.8 in which a number of principles are illustrated.

190 ORGANOMETALLIC COMPOUNDS
Figure 13.8. Analysis of alkene binding to di¨erent tricoordinated metal complexes.
The frontier orbitals of ethylene are shown on the left-hand side. On the right, occupying most of the ®gure, are ®ve sets of orbitals of the T-shaped complex. For clarity, only the a1 and b1 orbitals are shown since these can interact with the p …a1† and p …b1† orbitals of ethylene. The middle set is that of the PtClÿ3 complex (see also Figure 13.7). Lines a and b show the trend in d orbital energies as a function of the change of the metal, following Figure 13.2. Line b represents an average of the LUMO±HOMO gap, which was attributed to the trans e¨ect above. The separation between lines a and b is attributed to the ligand ®eld splitting parameter, D (or DO), which decreases in the series Pt > Pd > Ni. Lines c and d depict the corresponding changes with successive replacements of Clÿ by NH3. In this case, the downward slope of the fragment orbitals is due to increasing

AGOSTIC INTERACTION |
191 |
charge. There should be little di¨erence in the slopes of lines c and d since the metal center is the same. However, the HOMO±LUMO gap decreases since NH3 has a weaker trans e¨ect than Clÿ (see above). The interactions primarily responsible for the binding of the alkene are shown by bold dashed or dotted lines labeled i, ii, and iii. The attractive interaction, i, which is responsible for the majority of the binding energy, clearly is the weakest in Zeise's complex and increases in both series due to the slopes of lines b and d. At the same time, in both series, the attractive interaction, iii, decreases due to the slopes of lines a and c. In reference 283, the computed decrease in binding energy in both series was attributed to decreased p back donation in the series on the basis of the length of the CÐC bond. We note, however, that the length of the CÐC bond would also be increased by donation from the p bonding orbital, the primary bonding interaction in Figure 13.8 (or 13.7). On the basis of the diagram in Figure 13.8, it would be di½cult to blame the reduced binding to a further weakening of the already much weaker p back donation. It seems more likely that the culprit is the repulsive four-electron interaction, ii, which becomes more e¨ective in both series. Indeed, the di¨erence in the two series, namely that the binding energies are higher in the ligand substitution and lower in the metal substitution (relative to Zeise's complex), may be explained as due to the balance of interactions i and ii. Because of the constant HOMO±LUMO gap, interaction ii is relatively more important in the metal substitution series and has a greater e¨ect on the binding energy. In the ligand substitution series, the lower trans e¨ect of NH3 manifests itself as reducing the gap between the p orbital and the dx2 ÿ y2 LUMO, thereby increasing the primary interaction.
AGOSTIC INTERACTION
A common motif in organometallic chemistry is the agostic interaction, which can act to stabilize low-coordination low-e-count complexes. The requirement is an alkyl group with a b- or a g-CÐH bond attached to the metal within reach of (i.e., cis to) an empty coordination site. An attractive interaction occurs with the CÐH bond acting as a 2e donor into the low-lying metal valence orbital that occupies that site. In the case of a b-CÐH bond, hydride transfer may occur with little activation, resulting in an MÐH sigma bond and a p complex with an alkene as discussed above.
The reverse reaction corresponds to intramolecular CÐH bond formation. It requires that the alkene rotate from its equilibrium perpendicular orientation toward the less favored parallel orientation. The barrier hindering the rotation of the alkene may be partially or totally o¨set by the incipient agostic interaction. If the alkene is unsymmetrical, the question of regioselectivity of hydride transfer arises. In the case of an unsymmetrical alkene with an X: or ``C'' substituent, the donor p orbital is polarized away from the substituent (Figure 13.9) and the metal lies closer to that end. Conversely,

192 ORGANOMETALLIC COMPOUNDS
…a† |
…b† |
Figure 13.9. (a) Biased bonding in the p complex of a X:-substituted alkene; (b) regioand stereochemistry of intermolecular and intramolecular nucleophilic attack.
the acceptor p orbital is polaried toward the substituent. In this case, M-to-C hydride transfer occurs at the more highly substituted carbon.
It has been shown experimentally that attack by strong nucleophiles also occurs regioselectively at this C atom, stereoselectively from the face opposite to the metal [287]. Since the alkyl group s bonded to the metal is very carbanion-like, it is susceptible to protonation by acids, yielding an alkane. The overall reaction provides the mechanism for homogeneous hydrogenation of alkenes. It may be extended to hydrogenation of CÐN and CÐO pi bonds.
ZIEGLER±NATTA POLYMERIZATION
In the scheme above, the role of the hydride may very well be played by an alkyl group, particularly for the reverse reaction Figure 13.10 shows the basis for the most important industrial application of organometallic chemistry, the homogeneous polymerization of alkenes, modeled on the heterogeneous system ®rst discovered by Ziegler et al. [288]. The
Figure 13.10. A typical ole®n polymerization cycle, consisting of ole®n binding followed by MÐC rearrangement of alkyl group.

ZIEGLER±NATTA POLYMERIZATION |
193 |
catalytic systems consist of quasi-tetrahedral chloro or methyl complexes of d 0 |
metals |
with one or two cyclopentadienyl (Cp) ligands and a Lewis acidic aluminium-based cocatalyst whose function is to remove a ligand and produce the active metal catalyst, a coordinatively unsaturated complex [289]. Typically, the active metal catalysts are cationic species (e.g., Cp2TiMe‡ or Cp2ZrMe‡). For example, the following sequence based on Kaminsky and co-workers [290] yields high-density polyethylene:
Initiation with an alumoxane (large excess):
Cp2ZrMeCl ‡ …MeAlO†n ! Cp2ZrMe‡ ‡ Me…MeAlO†ÿn
Cp2ZrMe‡ ‡ CH2CH2 ! Cp2ZrMe…CH2CH2†‡
! Cp2Zr…CH2CH2Me†‡ …ˆCp2ZrR‡†
Propagation:
Cp2ZrR‡ ‡ CH2CH2 ! Cp2ZrR…CH2CH2†‡
! Cp2Zr…CH2CH2R†‡ …ˆCp2ZrR‡†
The electronic structure of such a d 0 complex (e.g., Cp2TiMe‡) is shown in Figure 13.11.
…a† |
…b† |
Figure 13.11. R ˆ alkyl, M ˆ a d 0 metal [e.g., Ti(IV) or Zr(IV)]: (a) cyclopentadienyl anion as a 2e donor ligand; (b) cyclopentadienyl as a 6e donor ligand.

194 ORGANOMETALLIC COMPOUNDS
Figure 13.12. Transition metal orbitals required for oxidative addition, a s-type acceptor and a p-type donor.
It may be derived from the ``D3h'' structure of Figure 13.4, in which both sets of degenerate orbitals are removed, leaving only an empty dz2 orbital. Such a complex is a weak Lewis acid, coordinating with the alkene through the latter's p bonding orbital. There are no occupied d orbitals to partake of p back bonding so the p orbital remains low and is receptive to transfer of the alkyl group, as in Figure 13.10. The nature of the transition structure and energetics of the alkyl transfer step (14 kJ/mol) have been studied by DFT [291]. The ``vacant'' site in the coordinatively unsaturated active catalyst is weakly occupied by solvent or by an intramolecular agostic interaction. A complete computational description of the intermediates and barriers involved in catalytic cycles of the type shown in Figure 13.10 has been carried out by Margl and co-workers [292].
OXIDATIVE ADDITION TO HÐH AND CÐH BONDS
Oxidative addition, reactions (13.1) and (13.2), was introduced in the opening paragraph of this chapter as one of the reactions of transition metal compounds of particular interest to organic chemists. Figure 13.12 shows a generic orbital interaction diagram for oxidative addition to H2. One notes that the coordination number of the metal increases by 2 in this process, as does the formal oxidation state of the metal. This e¨ectively limits the metal fragments which provide the occupied p donor (typically) dxz-type orbital and unoccupied s acceptor [usually …n ‡ 1†spzn or n dz2] orbital to 3-coordinated (Figure 13.4) and 4-coordinated (Figure 13.5) fragments. Examination of the fragment orbitals from Figure 13.4 reveals that a C3v fragment with s2d 2 (10e) con®guration and C2v fragments with s2d 4 (12e) or s2d 6 (14e) con®gurations are the most likely candidates
OXIDATIVE ADDITION TO HÐH AND CÐH BONDS |
195 |
since each has a LUMO suitable for interaction with a s bond donor and a HOMO which can act as a p donor. In the case of the s2d 6 C2v structure, this role would be ®lled by n dz2, and the midpoint of the incoming s bond will not be along the C2 axis but rather out of the plane of the metal's ligands. The 16e complexes of the type M(dmpe)2, dmpe ˆ (CH3)2PCH2CH2P(CH3)2, M ˆ Fe, Ru, have been shown experimentally and theoretically to add H2 essentially without activation [293]. Theoretical calculations on the model system, M(PH3)4, indicate that an initially formed complex between the metal LUMO ( pz of the C2v structure in Figure 13.5) and the HÐH bond coordinated endwise (h1-H2) collapses with little or no activation to an h2-H2 adduct as the metal HOMO (dxz of the C2v structure, Figure 13.5) comes into play. Reaction with methane (with Ru(CO)4) is predicted to follow the same pattern but with higher activation energy [294]. Oxidative addition of H2 to Fe(CO)4 has been shown experimentally to have an activation barrier less than 8 kJ/mol. Reductive elimination of H2 from Fe(CO)4H2 has an activation barrier of 86 G 9 kJ/mol. The average FeÐH bond dissociation energy (BDE) is 259 G 8 kJ/mol [295]. Computed MÐH and MÐC BDEs are available for CpMPH3R‡ complexes (M ˆ Co, Rh, Ir; R ˆ H, Me, Et, Pr) [296].

Orbital Interaction Theory of Organic Chemistry, Second Edition. Arvi Rauk Copyright ( 2001 John Wiley & Sons, Inc.
ISBNs: 0-471-35833-9 (Hardback); 0-471-22041-8 (Electronic)
CHAPTER 14
ORBITAL AND STATE CORRELATION DIAGRAMS
GENERAL PRINCIPLES
Orbital interaction theory as introduced and utilized in the previous chapters of the book has served to point us in a direction along a reaction coordinate. We have been able to ®nd the most favorable geometry in which two molecules can approach each other by considering the interactions of the frontier orbitals, and we have seen that, carried to the logical extreme, the initial interactions imply making and breaking of bonds and the formation of new molecules. Indeed, the initial trajectory implied from the orbital interaction theory very often does indeed lead to the expected products when the reaction is exothermic. For endothermic reactions, information with respect to the structure and stability of the transition state is necessary to decide upon the reaction pathway, and this could be deduced from examination of the reactive intermediates (or products if the reaction is concerted). The purpose of correlation diagrams is to follow a system continuously from one description to another, assuming that material balance is maintained, for example, a chemical reaction from reactants to products or a molecule from one conformation to another. A correlation diagram, or a comparison of several correlation diagrams, would permit one to identify preferred reaction pathways or more stable conformations and o¨er an explanation for experimentally observed preferences.
What should be correlated? In an orbital correlation diagram, the shapes and energies of orbitals are examined to see if the electronic structure of the reactants could be smoothly converted into the electronic structure of the products, each de®ned by the structures and occupancies of their respective orbitals. The nodal characteristics of orbitals are very resistant even to rather large perturbations and will tend to be conserved in chemical reactions. If an element of symmetry, for example, a mirror plane, is maintained during the course of the reaction, the nodal characteristics separate the orbitals into two sets, the members of one set being symmetric with respect to re¯ection
196
WOODWARD±HOFFMANN ORBITAL CORRELATION DIAGRAMS |
197 |
in the symmetry plane, the others being antisymmetric with respect to re¯ection. Orbital symmetries are conserved in the reaction. If the number of orbitals of each symmetry type is not the same in the reactants as in the products, that reaction will not take place readily. Even if symmetry is not present, local symmetry usually is su½cient to distinguish the orbitals, hence the principle of conservation of orbital symmetry which formed the title of the classic text by Woodward and Ho¨mann [3a] or the orbital correlation analysis using maximum symmetry (OCAMS) approach of Halevi [10].
WOODWARD±HOFFMANN ORBITAL CORRELATION DIAGRAMS
An abbreviated description of the generation of Woodward±Ho¨mann orbital correlation diagrams is presented here. A full description is available in numerous sources, most notably reference 3. To begin with, a reaction coordinate is proposed. The active components of the reactants and products are maximally symmetrized by ignoring substituents and the presence of embedded heteroatoms. Symmetry elements conserved in the course of the reaction (at least those which intersect bonds being formed or broken) are identi®ed, and group orbitals are combined to provide symmetry-adapted MOs if necessary. Inand out-of-phase combinations of like-symmetry reactant orbitals are taken if they are close in energy. The orbitals of products are treated in like manner. Inspection of the reactant and product orbitals may suggest obvious correlations. However, orbitals of like symmetry will not cross, while orbitals of di¨erent symmetry may. The lowest energy MO of the reactants is connected to the lowest energy product MO which has the same symmetry. The next higher reactant MO is similarly correlated with a product MO of the same symmetry type, and so on until the highest energy reactant MO has been correlated. The numbers and symmetry types of reactant and product orbitals must be the same, so that all orbitals are connected. If all reactant orbitals which are occupied in the ground-state electronic con®guration correlate with ground-state occupied MOs of the product(s), the reaction is thermally allowed by the postulated mechanism. If one or more reactant MOs correlate with product orbitals which are empty in the ground state, the reaction is not allowed.
Orbital correlation diagrams are useful for cycloadditions and electrocyclic reactions but not for sigmatropic rearrangements since no element of symmetry is preserved.
Cycloaddition Reactions
The orbital correlation diagrams for the attempted face-to-face dimerization of two ole®ns …p2s ‡ p2s† to form a cyclobutane and for the Diels±Alder reaction are shown in Figures 14.1a and 14.1b, respectively. Figure 14.1a displays a typical correlation diagram for a reaction which is orbital-symmetry forbidden. The HOMO of the ole®n pair (the out-of-phase combination of the two p bonds correlates with an unoccupied MO of cyclobutane. Therefore, an attempt to squeeze two ole®ns together in a face-to-face geometry (the least motion pathway to cyclobutane) would tend to yield a highly excited state of the cyclobutane. The reverse is also true. Rupturing a cyclobutane into two ole®ns would produce one or both ole®ns in a highly excited state. On the other hand, the orbital correlation diagram for the Diels±Alder reaction (Figure 14.1b) is typical of thermally allowed pericyclic reactions. Ground-state reactants would yield ground-state product(s) directly.
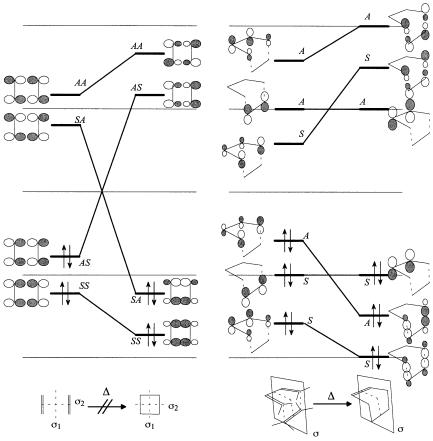
198 ORBITAL AND STATE CORRELATION DIAGRAMS
…a† |
…b† |
Figure 14.1. (a) Orbital correlation diagram for p2s ‡ p2s dimerization of two ole®ns to form a cyclobutane. (b) Orbital correlation diagram for p4s ‡ p2s cycloaddition of a diene and an ole®n (the Diels±Alder reaction).
Electrocyclic Reactions
As discussed in Chapter 12, electrocyclic reactions may proceed in a conrotatory or disrotatory fashion, that is, the p system cyclizes in an antarafacial or suprafacial manner, respectively. Since there is only a single component, it should be counted according to the general component analysis for a thermally allowed reaction, that is, p…4m†a ˆ conrotatory and p…4n ‡ 2†s ˆ disrotatory. Thus, the cyclization of butadienes to cyclobutenes (or more likely the reverse reaction) should proceed in a conrotatory fashion …m ˆ 1†, and the reaction of 1,3,5-hexatrienes should proceed in a disrotatory manner …n ˆ 1†. As illustrated in Figure 14.2, using butadiene as an example, a two-fold axis of symmetry is maintained throughout a conrotatory electrocyclic closure or opening, while a mirror plane of symmetry is preserved during disrotatory ring closure. The orbital correlation diagrams for electrocyclic reactions of butadiene and hexatriene are shown in Figures 14.3a and 14.3b, respectively. In each case, the solid lines connecting reactant and product MOs show the correlation for conrotatory ring closure, the MOs