

Drug Targeting Organ-Specific Strategies
.pdf8.5 Strategies to Deliver Drugs to Targets within the Tumour (Cells) |
207 |
208 8 Strategies for Specific Drug Targeting to Tumour Cells
(2)Expression of the antigen by normal tissues should be limited or, if the antigen is expressed on normal tissue, it should be inaccessible to antibodies in these tissues.
(3)The antigen should be membrane bound and not shed from the cell surface. One of the positive exceptions to this rule is carcinoembryonic antigen (CEA) which is also present in the serum of patients in significant concentrations.
The heterogeneity of tumours as well as the fact that their antigenic make-up resembles that of the equivalent normal tissues, has made it difficult to identify suitable target molecules. In the following, a number of potential target antigens for such an approach are discussed [23].The surface Ig idiotype sequences present in B-cell malignancies are close to ideal with respect to specificity as they truly represent a tumour specific antigen. However, antiidiotype targeting has several drawbacks that are difficult to overcome. First, the unique intrinsic specificity of the surface Ig implies that new antibodies have to be generated for every distinct B-cell clone. Second, soluble malignant B-cell-produced antibody present in the serum may act as a scavenger for the therapeutic anti-idiotypic antibodies thereby preventing them from binding to their membrane bound target [24]. Other B-cell-specific target antigens include the normal B-cell markers such as CD19 or CD20, which are present on a wide range of B-cell-derived malignancies. Immunotherapy directed against normal B-cell-specif- ic markers holds the risk of compromising the natural immune response by eradication of the complete B-cell repertoire. However it may be anticipated that this immune ‘gap’ can be restored by new, bone-marrow-derived B-cells.
Carcinomas are frequently occurring solid tumours. Examples of carcinoma-associated antigens that have been exploited in therapeutic protocols are c-erbB-1 or epidermal growth factor (EGF) receptor, c-erbB-2 or HERs/neu antigen, the folate receptor or folate-binding protein (FBP) and the epithelial glycoprotein-2 (EGP-2) [25]. Over-expression of the c-erbB-1 proto-oncogene product was reported in squamous cell carcinomas of the lung [26,27], adenocarcinomas and large cell carcinomas [28]. The proto-oncogene product c-erbB-2 is amplified in a variety of adenocarcinomas and squamous cell carcinomas, including lung, breast, gastric and colon cancer [29,30].The antigen is also expressed in normal lung tissue [28].
A number of both pre-clinical and clinical studies have used the folate receptor or FBP as a target for immunotherapy of ovarian carcinoma [31,32]. Expression of this tumour-associ- ated antigen by normal tissues is restricted [33]. The carcinoma-associated antigen, EGP-2, also called EpCAM, is a 38-kDA transmembrane glycoprotein, present on the majority of simple, stratified and transitional epithelia [34]. The biological function of EGP-2 has not yet been established.
Another approach in solid tumour therapy is to target antibodies to antigens expressed on the tumour vasculature, rather than to tumour-associated antigens of solid tumours. This has shown impressive activity in pre-clinical models [35,36]. Directing therapy to the accessible vascular compartment reduces the impact of the physical barriers of solid tumours, such as heterogeneous blood flow and elevated interstitial pressure [14]. Identification of appropriate target antigens that are expressed on the tumour vasculature, but not on cells of normal vessels, is an area of ongoing interest (see also Chapter 9).
Monoclonal antibodies against tumour-associated antigens or growth factors have been used to target the delivery of cytotoxic drugs, radionuclides and (bacterial) toxins [22]. Simi-
8.5 Strategies to Deliver Drugs to Targets within the Tumour (Cells) |
209 |
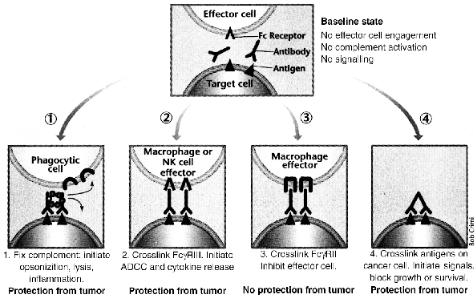
Figure 8.2. Monoclonal antibodies can block tumor growth using many mechanisms. Top, monoclonal antibodies recognize antigens on the target cell, in this case a cancer cell. 1. Monoclonal antibody bound to antigen activates complement components (small ring between the two antibody molecules), leading to opsonization of cancer cells by phagocytic cells expressing complement receptors (half-circles), direct lysis of tumor cells and inflammation with recruitment of inflammatory cells. 2. Monoclonal antibody binds to activating Fc receptors on the effector cells, leading to anti-body.dependent cellular cytotoxicity (ADCC) or release of cytokines. 3. Monoclonal antibody binds to inhibitory Fc receptors (or to both activation and inhibitory Fc receptors), inhibiting effector cell activation. 4. Monoclonal antibody binds directly to growth factor receptors or other signaling molecules on the cancer cell, leading to cell death reprinted with permission from [155].
larly cytotoxic immune effector cells have been redirected to kill tumour cells using bispecific antibodies [37]. These approaches will be discussed below.
8.5.1.2 Unconjugated Antibodies
Some unconjugated or ‘naked’ MAbs can induce anti-tumour effects by mechanisms that include the activation of the effector cells of the immune system, or the fixation of complement
(C) (Figure 8.2). The former, called antibody dependent cellular cytotoxicity (ADCC), depends on the ability of lymphocytes, macrophages, and granulocytes to recognize the Fc region (see Figure 8.1a) of the tumour cell-bound antibody.The latter involves activation of the C cascade that eventually punches holes in the plasma membrane of the target cell. Unfortunately, one of the inherent weaknesses of using mouse MAbs to treat humans is their inability to effectively activate human ADCC or human C because of structural differences between the Fc portions of mouse and human Igs [38]. Of the different subclasses of mouse
210 8 Strategies for Specific Drug Targeting to Tumour Cells
IgGs, IgG2a is the one which is most efficient in mediating human ADCC, whereas IgG3 can mediate potent C-dependent cytolysis [39].
Some MAbs have the ability to signal target cells to undergo cell cycle arrest (CCA) or apoptosis. The prototypic example of such a MAb is anti-Fas that signals apoptosis in all Faspositive cells [40,41]. However, because of the ubiquitous expression of Fas, administration of anti-Fas is lethal. Other MAbs, particularly when used as homodimers [42] which hypercrosslink their antigenic targets, can induce CCA or apoptosis. Both anti-CD19 and antiCD22 induce CCA in several Burkitt’s lymphoma cell lines both in vitro and in mice xenografted with human tumours [43]. Anti-Id MAbs are also thought to be of therapeutic value because of their ability to direct negative signals to tumour cells [44].
More recently, our knowledge of cellular signalling pathways has led to the development of MAbs which target molecules involved in the regulation of tumour cell growth. Cytostatic or cytotoxic effects can result from the binding of a MAb to growth factors or cellular growth factor receptors which are required for tumour cell survival [45]. For example, many adult carcinomas depend, in part, on the autocrine or paracrine effects of epidermal growth factor (EGF) or transforming growth factor-α (TGF-α).As a result, some anti-EGF receptor MAbs have anti-tumour activity in tumours of the breast, vulva, cervix, and in squamous cell carcinomas [45]. Other MAbs targeting various cell surface growth factor receptors have also effectively induced CCA or apoptosis in tumour cells [40,46,47].
To potentiate the cytotoxic effects of MAbs with low endogenous activity, cytokines and activated effector cells have been co-administered [48]. Cytokines can increase extravasation of MAbs into the tumour and, by inducing local inflammatory responses, enhance the influx of effector cells. For example, the addition of interleukin-2 (IL-2) or the concomitant adoptive transfer of lymphokine-activated killer cells (LAKs) can enhance the activity of MAbs. Other cytokines, such as interferon-gamma (IFNγ) and IFNα can augment the delivery of MAbs to tumour targets by upregulating antigen expression [48,49]. The use of activated effector cells (peripheral blood mononuclear cells or granulocytes) in combination with MAbs has also resulted in their increased cytotoxicity to various tumours [48].
8.5.1.2.1 Potential Disadvantages and Limitations of the MAb Approach
Unfortunately, the clinical efficacy of MAb-directed therapy is often limited. One important factor in this respect is that the target antigen is expressed on normal as well as malignant cells. With the exception of MAbs to idiotypic domains of B-lymphocytes, MAbs which are exclusively tumour-specific have not been identified. Rather, most currently used MAbs recognize tumour-associated antigens expressed at higher density on malignant cells relative to normal cells. Furthermore, MAbs are murine in origin, particularly those used in past research. As a consequence, human anti-mouse antibody (HAMA) responses developed in patients treated with murine MAbs, led to accelerated clearance of the administered MAb and blocking of the therapeutic effect.
As mentioned in Section 8.4.2, elevated interstitial pressure, heterogeneous and reduced functional vasculature, and the relatively large distances that Mabs have to travel in the tumour interstitium, are hurdles which need to be overcome in the pursuit of efficient drug targeting. The relatively large molecular weight of Mabs (approximately 150 kDa) [2,14], also
8.5 Strategies to Deliver Drugs to Targets within the Tumour (Cells) |
211 |
contributes to limited tumour penetration and minimal efficacy especially if MAb-directed therapies are used as single agents in patients with advanced disease.
Several modifications have been explored to improve efficacy. The problem of relatively large molecular weight can be partially resolved by using fragments of IgG generated by enzymatic digestion (Figure 8.1b). With advances in protein engineering, efforts are being aimed at reducing the size of the MAb, as well as reducing immunogenicity by using chimeric or humanized MAbs [39] (see Section 8.5.1.3). Despite these concerns, adverse effects of naked MAb, even after repetitive administration, are uncommon, and when they occur they are usually readily reversible.
8.5.1.3 Recombinant Antibodies
8.5.1.3.1 Recombinant DNA Technology
Recombinant DNA technology can be exploited to deal with the above-mentioned problems and has been used not only to manipulate the size, but also the shape, affinity, and immunogenicity of the MAb molecule. Chimeric versions of murine MAbs can be constructed through combination of variable chains of the original murine MAb with the constant domains of human Ig. This serves to enhance effector functions and reduce the chances of a HAMA response occurring (Figure 8.1c). Alternatively, the six hypervariable loops (complementarity determining regions, CDRs) forming the antigen binding site of a murine antibody can be transplanted into a human framework resulting in a CDR-grafted or humanized antibody (Figure 8.1c).
8.5.1.3.2 Single Chain Fv Antibody Fragments
In addition to modified complete Ig molecules, recombinant DNA technology has been used to construct small antibody-like molecules called single chain Fv fragments (scFv) [50,51] (Figure 8.1c). Briefly scFvs are recombinant antibody fragments consisting of only the variable light chain (VL) and variable heavy chain (VH) domains covalently connected by a flexible polypeptide linker typically composed of 15 amino acid residues ((Glycine4Serine)3). Due to their relatively small size (approximately 26 kDa), scFvs are rapidly distributed and a significant improvement in penetration into solid tumours has been shown in vivo. Murine scFv fragments can be produced by PCR-based gene assembly using mRNA templates isolated from the corresponding hybridoma cell line [52].
Functional expression (display) of scFv proteins on the surface of bacteriophage has been widely exploited in the selection of scFvs which have retained the binding properties characteristic of the MAb from which they were derived (see also Chapter 10 on the application of phage display for target antigen-specific scFv identification). The inherent advantage of phage display technology is its direct link between the DNA sequence and the protein function [53]. Large numbers of clones can be rapidly screened for antigen binding, making it the method of choice for hybridoma Ig cloning. It is clear that molecular cloning and sequencing of scFv forms the basis for further antibody engineering and modelling.
212 8 Strategies for Specific Drug Targeting to Tumour Cells
8.5.1.3.3 Phage Display Library
As an alternative to immunization and hybridoma construction procedures, it is possible now to construct large (synthetic) human antibody gene repertoires entirely in vitro (see also Chapter 10). This procedure can generate a huge library of recombinant filamentous bacteriophages that express hundreds of millions of different human scFvs on their tips fused to the phage minor coat protein III [38,54]. The scFvs displayed by these phages can show antigen binding activity and phages with the desired binding characteristics and specificity can be selected by panning on the antigen. The selected phage (including the genetic information of the displayed scFv inside) can be rescued and grown after each round of panning after which the ‘enriched’ phage library is again subjected to selection so that even rare phages (< 1/108) can be isolated. Using this strategy human antibodies and/or their fragments have been isolated with specificities against foreign and self antigens [55].
8.5.1.3.4 Transgenic ‘Human’ Animals
A further advance in antibody technology is the development of transgenic mouse ‘human’ strains. XenoMouse animals have been engineered in such a way that they now produce exclusively human antibodies rather than murine antibodies when immunized. The use of XenoMouse animals to produce MAbs avoids the need for any engineering of the antibody genes, since the products are already 100% human protein. XenoMouse animals are fully compatible with standard hybridoma technology and can be readily adopted by laboratories experienced in monoclonal antibody production [56].
8.5.1.3.5 Considerations for Recombinant Antibody Production
When the biodistribution of scFv (Figure 8.1c), Fab′, (Fab)2′ (Figure 8.1b), and IgG (Figure 8.1a) were compared, most of the intact IgG delivered to tumours was concentrated in the region immediately adjacent to the blood vessels.The Fab′ and F(ab)2′ fragments demonstrated intermediate degrees of tumour penetration, while the scFv was distributed more evenly throughout the tumour [57].
In mice with human breast carcinoma xenografts, a humanized IgG anti-HER-2 MAb eradicated well-established tumours [58]. In addition, a humanized version of an IgG antiCD33 MAb (HuM 195) mediated ADCC in vitro [59] and had an 8.6-fold higher avidity than the parent murine Mab. Recombinant antibody fragments may have valuable properties as discussed above, but their biophysical behaviour, production yield and low thermostability leaves much to be desired and thereby limits their usefulness for in-vivo applications so far [60]. One possibility to improve these characteristics of scFv fragments with suboptimal stability and/or folding yield, is the grafting of their CDRs onto the framework of a different, more stable scFv [61,62].
Another valuable tool for the development of scFv-based therapeutics consists of a versatile expression vector for the rapid construction and evaluation of scFv-based fusion proteins and bispecific scFv [63]. The vector was used for grafting a number of biological effector princi-

8.5 Strategies to Deliver Drugs to Targets within the Tumour (Cells) |
213 |
ples onto anti-EGP-2 scFv. Biologically-active fusion proteins were produced by directing them through the endoplasmic reticulum-based protein folding machinery of eukaryotic cells. This procedure may help to identify those fusion proteins that which desirable characteristics such stability and biological activity in the presence of serum and at low protein concentrations.
Biophysical properties such as high thermal stability are thus of paramount importance in the decision as to whether or not these molecules are useful in vivo. The above described approaches may provide a strategy to meet these requirements and may eventually result in attractive modalities for the targeting of solid tumours in patients.
8.5.1.4 Immunotoxins (ITs)
The conjugates referred to as ITs are hybrid molecules consisting of MAbs linked to powerful toxins (or toxin subunits) purified from plants, fungi, or bacteria [64] (Figure 8.1e and Table 8.1). These toxins inhibit protein synthesis after internalization, leading to death of the targeted cell. Small quantities of ITs when compared with unconjugated MAbs, are needed for effective target cell killing. In fact, a single toxin molecule in the cytosol can kill a target cell, and, unlike chemotherapeutic agents, ITs will kill both resting and dividing cells.
Limitations to IT therapy include their immunogenicity and toxicity. Dose-limiting side-ef- fects of IT therapy include hepatotoxicity and vascular leak syndrome.
Table 8.1. Toxins ued for the preparation of ITs.
Source |
Toxin |
Enzymatic activity |
|
|
|
Plant |
Type I RIPs* (single chain) |
N-glycosidase for 28s rRNA |
|
Pokeweed anti-viral protein (PAP) |
|
|
Saporin (SAP) |
|
|
Gelonin |
|
|
Momordin |
|
|
Trichosanthin |
|
|
Barley toxin |
|
|
Type II RIPs (two chains) |
|
|
Abrin |
|
|
Ricin |
|
|
Viscumin |
|
Bacteria |
Diphtheria toxin (DT) |
ADP Ribosylation of EF2 |
|
Pseudomonas exotoxin (PE) |
|
Fungi |
α-Sarcin |
Ribonuclease for 28 S RNA |
|
Restrictocin |
|
|
|
|
* RIP: Ribosome-Inactivating Proteins
8.5.1.5 Monoclonal Antibody–Drug Conjugates
MAb–drug conjugates offer the advantages of improving the therapeutic index by increasing drug uptake by tumour cells, reducing drug toxicity to normal cells, and prolonging bioavailability of the drug and thus more extensive exposure to tumour cells.

214 8 Strategies for Specific Drug Targeting to Tumour Cells
Conventional cytotoxic drugs such as doxorubicin, idarubicin, bleomycin, methotrexate, cytosine arabinoside, chlorambucil, cisplatin, vinca alkaloids, and mitomycin C have all been conjugated to tumour-binding MAbs [65–69] (Figure 8.1e). Drug conjugates can be prepared by covalently coupling drugs directly to a MAb or indirectly through an intermediate spacer molecule such as dextran, human serum albumin, poly-glutamic acid, carboxymethyl dextran, or amino-dextran. An indirect linkage facilitates the attachment of more drug molecules to one MAb molecule, in theory resulting in an increased delivery of drug molecules to the tumour (see Chapter 11 on drug–carrier conjugate synthesis strategies).
The members of the enediyne family of antibiotics are highly potent drugs that are good candidates for attachment to MAbs [70]. Calicheamicin conjugates of e.g. the MAb CT-M-01, exerted strong cell specific activity against s.c. breast-tumour xenografts in athymic mice [71]. Similar impressive anti-tumour activity was shown with a calicheamicin conjugate of anti- ganglioside-GD2 MAb used to treat experimental liver metastases in immunocompetent mice [72].
Table 8.2. MAb-drug conjugates that have been developed for cancer therapy.
Class of anti-neoplastic drug |
Drug |
Disease |
|
|
|
Antimetabolites |
Methotrexate |
Lung cancer, Colon cancer, |
|
|
Teratocarcinoma, T cell |
|
|
lymphoma |
|
5-Fluorouracil |
B leukaemia |
|
Cytosine arabinoside |
B leukaemia |
|
Aminopterin |
Murine thymoma |
|
5-Fluoro-2’-deoxyuridine |
Colon carcinoma |
Alkylating agents |
Chlorambucil |
Murine thymoma, Murine |
|
|
lymphoma, Melanoma |
|
Melphalan |
Colon Cancer, Murine thymoma |
|
Mitomycin C |
Lung cancer, Various dissemina- |
|
|
ted refractory malignancies, |
|
|
Gastric cancer |
|
Cisplatinum |
Ovarian carcinoma |
|
Trenimon |
Hepatoma |
Anthracyclines |
Doxorubicin/adriamycin |
Melanoma, Ovarian carcinoma, |
|
|
T-cell lymphoma, Colon |
|
|
carcinoma, B-cell lymphoma, |
|
|
Various disseminated refractory |
|
|
malignancies, Breast cancer, Lung |
|
|
cancer, Pancreatic cancer, Liver |
|
|
cancer, Neuroblastoma |
|
Daunomycin |
Soft-tissue sarcomas, Mammalian |
|
|
carcinoma, Hepatoma |
Antimitotic agents |
Vinca alkaloids |
Lung adenocarcinoma |
Miscellaneous agents |
Bleomycin |
Leukaemia |
|
Idarubicin |
Murine thymoma |
|
Maytansine |
Colon cancer |
|
Calicheamicins |
Human breast |
|
|
carcinoma xenografts |
|
|
|

8.5 Strategies to Deliver Drugs to Targets within the Tumour (Cells) |
215 |
In the case of drug–monoclonal antibody conjugates, the entire conjugate may be internalized after which the drug can be released intracellularly. The drug may also be cleaved extracellularly and subsequently taken up into tumour cells by diffusion or active transport. Success with drug–MAb conjugates has been limited thus far because of poor uptake of the MAb–conjugates especially in solid tumours. Drug delivery is also limited by the number of drug molecules that can be efficiently carried by each antibody molecule. Furthermore chemical conjugation is usually a complex procedure that can damage both the MAb and the drug and MAb–conjugates require intraor extracellularly active biochemicals and/or enzymes to cleave the active drug from the antibody. Table 8.2 gives an overview of the various anti- body–drug conjugates that have been developed for cancer therapy to date.
8.5.1.6 Radioimmunoconjugates
Another way of using MAbs as therapeutic agents is to couple them to radionuclides (Figure 8.1e and Table 8.3). Radioimmunoconjugates offer many advantages in the treatment of cancer. Cell killing does not rely on the host’s immune system and occurs by the ionizing effects of emitted radioactive particles [73]. These radioactive cytotoxic particles are effective over a distance of several cell diameters, allowing eradication of antigen-negative cells by the radioimmunoconjugate bound to the adjacent antigen-positive tumour cells. This is useful considering the heterogeneity of antigen expression in some tumours. Finally, the amount of radioactive MAb delivered to a tumour can be measured non-invasively by imaging [67].The most important factors for therapeutic efficacy of radioimmunoconjugates are good penetration, favourable pharmacokinetics, and a prolonged time of retention in the tumour [74].
Table 8.3. Isotopes used for radioimmunotherapy in cancer
|
Radioisotope |
|
|
Beta-emitters |
lodine-131 |
|
Yttrium-90 |
|
Rhenium-188 |
|
Rhenium-186 |
|
Copper-67 |
Alpha-emitters |
Bismuth-212 |
|
Astatine-211 |
Electron capture |
Iodine-125 |
8.5.2 Bispecific Monoclonal Antibodies
Another approach to selectively inducing tumour cell killing is by the use of bispecific monoclonal antibodies (BsMAb). They combine the specificity of two separate antibodies within one molecule and cross-link an effector killer cell or a toxic molecule with the target cell to be destroyed [75]. There are three major approaches for creating BsMAbs. They can be obtained by chemical cross-linking of two MAbs, by fusing two hybridomas [76], or by genetic

216 8 Strategies for Specific Drug Targeting to Tumour Cells
engineering [77]. Each method has its advantages and disadvantages. Chemical conjugates have a well-defined linkage and can be produced in high yield. However, there is lot-to-lot variability in purity and activity. Quadromas, produced by fusing two hybridomas, can also produce large quantities of BsMAbs. However, in addition to the desired BsMAb, the parental MAb and every possible combination of heavy and light chain matches and mismatches are also produced. Furthermore, quadromas are often genetically unstable and require frequent subcloning. With recombinant fusion proteins, it is possible to make new combinations of Fab or Fv segments or to combine human and mouse gene segments. Yields and correct folding of the purified fusion protein can present problems as discussed earlier.
Cytotoxic drugs including toxins such as saporin, ricin A chain, vinca alkaloids, and radioisotopes have been delivered to tumour cells with BsMAbs that bind to the drug/toxin with one arm and to a surface molecule on the targeted cell with the other arm.This approach has proven successful in animals as e.g. shown by Schmidt et al. [78].
Cytotoxic effector cells have also been cross-linked to tumour cells via BsMAb (Figure 8.3). The BsMAb activates the cytotoxic activity of the effector cell on bridging it to the target cell. Several effector cells, including phagocytic cells, natural killer (NK) cells and T lymphocytes, can mediate cellular cytotoxity [37,75,79,80].Adequate pre-activation of the effector cells is an important requirement in these methods of drug delivery. In the case of T
Figure 8.3. Schematic representation of bispecific antobody mediated tumor cell recognition by an immune effector cell. Summarised are effector cell types, trigger molecules and tumor associated antigens used as a targed as reported in the literature. From reference [37].