

Supplement F2: The Chemistry of Amino, Nitroso, Nitro and Related Groups.
Edited by Saul Patai Copyright 1996 John Wiley & Sons, Ltd.
ISBN: 0-471-95171-4
CHAPTER 21
Displacement and ipso- substitution in nitration
J. P. B. SANDALL
Department of Chemistry, University of Exeter, Stocker Road, Exeter EX4 4QD, UK Fax: (+44) 1392 263434; e-mail: J.P.B. Sandall@exeter.ac.uk
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
949 |
II. FORMATION OF ipso-ADDUCTS . . . . . . . . . . . . . . . . . . . . . . . . . . |
950 |
III. FURTHER REACTIONS OF ipso-INTERMEDIATES |
|
AND ADDUCTS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
957 |
A. Solvolysis of Adducts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
957 |
B. Further Addition to the ipso-Species . . . . . . . . . . . . . . . . . . . . . . . |
961 |
IV. REARRANGEMENT OF ipso-SUBSTITUTED GROUPS . . . . . . . . . . . |
964 |
V. DISPLACEMENT OF ipso-SUBSTITUTED GROUPS . . . . . . . . . . . . . |
969 |
VI. CONCLUSION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
970 |
VII. REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
970 |
I. INTRODUCTION
This review covers the literature from about 1980, the year of publication of Schofield’s1 book on nitration which includes a chapter on ipso-attack in nitration. The use of the term ipso dates back to 1971 when Perrin and Skinner2 applied it to systems undergoing attack at a substituted position in an aromatic ring. This process has always been regarded as unusual in electrophilic processes such as nitration, since the requirement of a positively charged leaving group results much more often in loss of a proton than loss of a less stable positively charged species. However, this view is partly a consequence of seeing the nitration reaction only as a method of introducing the nitro group; when one considers all the possible reactions that might result from attack at an ipso-position, then it is clear that the process is of considerable importance. In nucleophilic substitution of course ipso-substitution is the norm, since the reversed polarity of the reaction results in loss of a stable negatively charged substituent (e.g. halide ion) much more commonly than a hydride ion, although this latter process can be competitive in special circumstances. Another reason for the greater attention paid to reaction at an ipso-position recently is that modern analytical techniques enable the characterization of less stable or transient
949

950 |
J. P. B. Sandall |
species, or compounds only present in small amounts, leading to a much fuller description of complicated mechanisms.
The general consequences of ipso-attack in a nitration reaction may be briefly outlined. Usually the aromatic substrate will have two substituent groups, one of which, not necessarily the more electron releasing, will direct an incoming electrophile to attack at a substituted position; thus, for example, 1,4-dimethylbenzene will react with nitronium ion, in a sufficiently acidic medium, to generate the 1,4-dimethyl-1-nitro-benzeneonium ion. For 4-chloroanisole, the dominant ipso-products will arise from attack at the position bearing halogen. This positively charged species may react to form a more stable species in several ways, for example by losing a proton from a suitable position, e.g. formation of 4-methyl-4-nitro-cyclohexa-2,5-dienone (often called an ipso-intermediate) from 4-methylphenol as starting material. Alternatively it may be stabilized by capture of a negatively charged species usually derived from the solvent, e.g. acetate ion from acetic acid, usually called an ipso-adduct. The ipso-species formed by either of these routes may dissociate again (possibly homolytically, generating nitrogen dioxide); either the ipso- substituent or the nitro group may rearrange (conceivably either homoor heterolytically) resulting in either nuclear or side-chain substitution or it may undergo solvolysis. Other reactions include further addition to its carbon carbon double bonds. Finally, a suitable substituent, e.g. iodo, may simply be displaced by the incoming electrophile. It must also be remembered that the substrates suitable for ipso-attack are often among the most reactive; this frequently gives rise to the possibility of nitrous acid catalysed nitration as a further complication or even direct reaction with nitrogen dioxide. Examples illustrating all these types of behaviour have been recently investigated.
II. FORMATION OF ipso-ADDUCTS
Major work in this field has been carried out by Fischer, Henderson and their coworkers3 12. The behaviour of 1,4-dimethylbenzene on nitration may be taken as a typical example4. This substrate, when nitrated in acetic anhydride, gives rise to a diastereomeric pair of adducts involving the formal addition of nitronium acetate across the 1,4-positions (1). In earlier work, attempts were made to assign the configuration of such adducts by using proton NMR shift reagents, but with only limited success. However, relative configurations of the acetates, and dienols derived from these, had been obtained. Myhre and coworkers13 had attacked a similar problem by synthesizing 1,4-dialkyl-4- nitrocyclohexadienols by addition of methyllithium to a 4-alkyl-4-nitrocyclohexadienone. This reaction is somewhat stereoselective, and one would expect the predominant isomer to have (Z) configuration, since addition trans to the nitro group is preferred. This is the isomer which (as acetate) is formed in smaller amount in the nitration process. Fischer and coworkers prepared the two diastereoisomeric 1,4-dimethyl-4-nitrocyclohexa-2,5-dienyl acetates and separated them by low temperature chromatography (overall yield 90%, ratio approximately 7:3). The acetates were stereospecifically reduced to the corresponding dienols (aluminium hydride) and these, again stereospecifically, were methylated to give the ethers. The 2,6-dideuterio-1,4-dimethyl-4-nitrocyclohexadienols were prepared using the method of Myhre and coworkers, separated, and the 13C NMR spectra assigned for both isomers of the alcohol, the ether and the acetate. Comparison with the spectra of the isomeric acetates derived from direct nitration allowed the assignment of the major isomer to the (E) configuration (2). Sufficiently good crystals of the (E)-1,4-dimethyl- 4-nitrocyclohexa-2,5-dienol were obtained to carry out an X-ray structure analysis; the cyclohexadiene ring is almost flat with the methyl groups trans as expected. It may be noted that the stereochemistry of these acetates and ethers is wrongly assigned in an earlier paper by Shosenji and coworkers14.
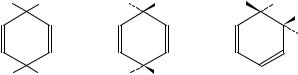
|
21. Displacement and ipso-substitution in nitration |
951 |
||||
Me |
NO2 |
Me |
NO2 |
Me |
NO2 |
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
OAc |
AcO |
Me |
AcO |
Me |
|
|
|
|
(1) |
(2) |
|
|
(3) |
|
Whilst aromatic compounds substituted 1,4- with alkyl groups will give mainly 1,4- ipso-adducts by nitration at low temperatures, a complication arises with many other substituents, e.g. a halogeno group para to alkyl. This is the possible formation of 1,2- adducts (3), as well as the normal 1,4-addition observed with 1,4-dialkylbenzenes (except when one group is t-butyl). Thus nitration of 4-fluoromethylbenzene in acetic anhydride at low temperature10 gives the expected pair of diastereomeric 1,4-nitronium acetate adducts, but in addition the cis-1,2-adduct is observed. In this case the ratio of 1,4-addition to 1,2- addition is about 10:1. However, replacement of the 4-fluoro-substituent by 4-chloro or 4-bromo results in only formation of the 1,2-adduct being observed, with the nitro and acetate groups again being cis to each other. In both cases, the NO2 group is attached to the activated ipso-methyl position and not the ipso-halogen position. This behaviour may be rationalized by the relative activations of the ipso and unsubstituted positions towards the electrophile using simple additivity of the partial rate factors for the appropriate monosubstituted benzenes including the rate factors for the ipso positions. The nitro and acetate groups in these 1,2-adducts are always cis; the authors conclude that the addition is stereoselective, being in all cases syn addition. This was confirmed by an X-ray crystal structure determination in the case of the 4-bromo product. Presumably the ipso- intermediate carbocation does not escape from the substrate/nitronium acetate encounter pair. The authors also comment on the interesting regioselectivity mentioned above: with both 1,4-dimethylbenzene and 4-fluoromethylbenzene the addition is effectively 1,4; for the other 4-substituents investigated (chloro, bromo, methoxy) addition is almost completely 1,2. This similarity in behaviour for fluoro and methyl does not parallel either the inductive or resonance (or total) electronic effects of the substituents on an electrophilic process.
In an attempt to investigate further this regioselectivity, the group of Fischer and Henderson11 looked at the nitration of a series of 2- and 3-substituted 4-methylanisoles and phenols. The product distribution by and large followed the expected pattern calculated from the partial rate factors for halogen and nitro substituents on the observed ratios of attack on 4-methoxymethylbenzene. The largest deviations from the expected regioselectivity occurred with a nitro group in the 2-position: it was expected that this should slightly increase ipso-attack; in fact it significantly decreased it, possibly due to steric effects. However, an examination of the relative amount of capture of the presumed 4-methoxy ipso-cation at the 2-, 4- and 6-positions showed some interesting anomalies. The extent of 4-capture versus 2/6 is again not related to the electronic effects of the substituents in the 3-position, nor is the fact that selectivity between the 2- and 6-positions reversed by exchanging a 3-chloro for a 3-nitro substituent easily explained. Although the grouping of fluoro and methyl substituent effects referred to above suggests the possibility of a radical process, the marked sensitivity of the reactions to substituent effects generally would seem to preclude this.
Whereas capture of the ipso-cation by acetate in acetic acid solution continues to be one of the best methods of isolating such adducts, its capture by water is, of course, of great importance during normal nitration in aqueous acid. For example, in 60% H2SO4 1,2- dimethylbenzene gives no less than 33% of monoand di-nitro-3,4-dimethyl phenols15.
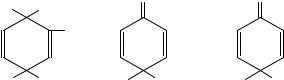
952 |
J. P. B. Sandall |
The general process is one of loss of nitrous acid from the adduct (4); in highly substituted aromatics the initially formed adduct may well also rearrange either by hydroxyl or other group migration.
The second major route to ipso-species formation may be formulated as loss of a proton (generally from a hydroxyl or amino group) from the initially formed cation. For example, in the nitration of 2,6-dichloro-4-methylphenol, the major initial product was shown to be 4-methyl-4-nitro-2,6-dichlorocyclohexa-2,5-dienone16 as early as 1900. A more recent example, investigated by Ridd and coworkers17, is of formation of ipso-intermediates during the nitration of N,N-dimethyl-p-toluidine and some related compounds. The authors showed that the major product from the toluidine itself (2-nitro- N,N-dimethyl-p-toluidine, 78%) was formed in two stages; the intermediate was clearly demonstrated by both 1H and 13C NMR to be the ipso-adduct (5). The marked upfield shifts of the 4-methyl group protons (0.3 ppm) and the methyl carbon itself (38 ppm) are completely in accordance with the change from sp2 to sp3 hybridization at this position. The spectrum of the corresponding 4-ethyl-N,N-dimethylaniline also showed the same phenomenon as did N,N-2,4,6-pentamethylaniline where the alternative possibility of an ortho ipso-intermediate was shown to be absent. The solvent used was a typical nitrating mixture (HNO3 in 70% H2SO4) and the intermediates had half-lives up to several hours at 0 °C. The clean separation of the 1H NMR peaks for the 4-methyl group in starting amine, intermediate and the final product enabled a kinetic study to be carried out on the formation of both intermediate and product. This showed that formation of the intermediate was an autocatalytic reaction, inhibited by hydrazine and strongly catalysed by traces of nitrous acid. It is clear therefore that the intermediate is formed not by nitronium ion attack at the 4-position, but by a process involving catalytic quantities of nitrous acid, which can be formed by side-reactions. This kinetic behaviour parallels exactly that of N,N-dimethylaniline18 which, under the same conditions, gives almost exclusively N,N,N,N-tetramethylbenzidines by dimerization of the cation radical PhNMe2Cž which is formed by electron transfer to the NOC ion. The possible alternative mechanism, nitrosation at the 4-position followed by oxidation, was rejected on the grounds that benzidine formation could not be explained. Further confirmation of the oxidizing function of nitrous acid (NOC at this acidity) is given by the observation during the reaction of the ESR spectrum of an organic free radical shown by simulation to be the N,N-4-trimethylanilinium cation radical. Although the authors noted at the time that the observation of the radical did not prove it was on the reaction path, later work showed that the intermediate and final nitro product exhibited a CIDNP effect which confirmed the postulated mechanism. Formation of the ipso-product by NO2C attack also occurs at appropriate acidities. The rearrangement step will be considered later.
|
|
|
+ |
|
+ |
Me |
NO2 |
|
NMe2 |
|
NMe2 |
|
|
|
|
||
|
|
CH3 |
|
Me |
Me |
HO |
H |
NO2 |
Me |
NO2 |
Me |
(4) |
|
|
(5) |
|
(6) |
The example outlined above is a particularly clean reaction; most ipso-attacks in amines and phenols are rarely as simple as this. Two other substrates might be used as examples
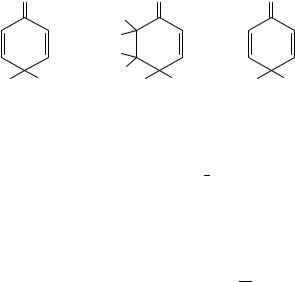
21. Displacement and ipso-substitution in nitration |
953 |
at this point: N,N-2,4,6-pentamethylaniline and N,N-2,4-tetramethyl-6-nitroaniline19. At 0 °C in 70% HNO3 the former compound undergoes ipso-attack at the 4-position to form initially the relatively stable intermediate (6). This can be crystallized out of solution as a hexafluorophosphate, or, if the acid solution is somewhat diluted (40%), it can capture water to generate the 2,4,6-trimethyl-4-nitrocyclohexa-2,5-dienone (7) by the slow displacement of the dimethylamino group. On the other hand, substitution of the 6-methyl group by 6-nitro leads to a completely different final product. Here the analogous ipso- intermediate is seen only as a transitory species; the major product (8) may be formulated as arising by the formal addition of one molecule of water across the 5,6 double bond in the ipso-intermediate corresponding to 6.
|
|
|
+ |
|
|
O |
|
|
NMe2 |
|
O |
Me |
Me |
NO2 |
|
|
|
H |
|
|
|
||
|
|
|
|
|
|
|
|
H |
|
|
|
|
NO2 |
HO |
NO2 |
|
NO2 |
Me |
Me |
Me |
|||
(7) |
|
|
(8) |
|
(9) |
The nitration of 4-methylphenol provides an interesting example of how the existence of a reasonably stable ipso-intermediate (9) modifies the nitration process and also is markedly affected by the exact reaction conditions. Coombes and coworkers20 investigated products and kinetics for this reaction using HNO3 in 60 80% H2SO4. The amount of the expected product, 2-nitro-4-methylphenol, although approaching 100% at high acidities, was much less at lower acidities unless a more efficient nitrous acid trap (sulphanilic acid) was used than the sulphamic acid, which was the trap required for the kinetic studies. The results of the kinetic study showed that the ipso-intermediate (9) was formed by a process first order with respect to nitric acid, and that it decomposed by an overall first order process at a rate (although dependent on overall acidity) independent of nitric acid concentration. In the presence of nitrous acid catalysis, presumably, a much greater fraction of the reaction went by the ipso-intermediate route other reactions of this species would reduce the yield of the final nitro product.
This reaction was subsequently investigated21 in acetic anhydride as solvent, using nitronium acetate as the nitrating agent. Under these conditions nitrous acid catalysed nitration is the almost exclusive route to both the ipso-intermediate (30%) and the 2- nitro-4-methylbenzene (70%). The temperature at which the ipso-compound was formed was sufficiently low to preclude its rearrangement to the final nitro product. Both 15N and 13C CIDNP effects were seen. A strong emission signal in the 15NMR spectrum corresponding to the ipso-nitro compound (9) was observed, as were an enhanced 13C NMR absorption signal for C-4 and an emission signal for C-1. The starting material also showed a CIDNP effect in its 13C spectrum, the reverse of that for the intermediate, i.e. the C-4 signal in emission and the C-1 signal in enhanced absorption. In order to interpret these effects, Kaptein’s rules22 were applied, using the known g values for NO2 and the 4-methylphenoxy radical; the signs of the necessary hyperfine coupling constants were derived for C-1 and C-4 from semi-empirical molecular orbital calculations. The agreement between the observed phase of all the polarized signals and that deduced from the mechanism (Scheme 1) is complete. Thus, the results are only consistent with a mechanism in which the polarization of the ipso-intermediate arises from diffusion
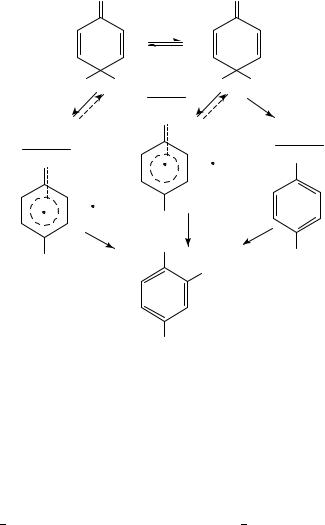
954 |
|
J. P. B. Sandall |
|
|
|
|
|
|
+ |
|
|
|
O |
|
OH |
|
|
|
|
H+ |
|
|
|
Me |
NO2 |
|
Me |
NO2 |
|
|
|
|
|
Path C |
|
|
Path A |
OH |
Path B |
|
|
|
|
|
|
|
|
O |
|
+ |
NO2 |
OH |
|
|
|
|
|||
|
|
|
|
||
NO2 |
|
|
|
NO |
+ |
|
Me |
|
2 |
||
|
|
|
|
||
|
|
OH |
|
Me |
|
Me |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
|
Me
SCHEME 1
together with the 4-methylphenoxy radical and NO2, not by direct electron transfer to the nitronium ions, nor from the radical pair generated by electron transfer to the nitrosonium ion. Also, the not inconsiderable polarization found in the substrate shows that there must be a significant probability of escape from the radical pair (Path A, Scheme 1) and that the initial oxidation step must also be reversible. This reversibility in the primary oxidation step had already been deduced from a kinetic study23 under quite different conditions, i.e. for nitrous acid catalysed nitration in aqueous nitric acid. The subsequent rearrangements (Paths B and C) will be dealt with later.
Formation of ipso-intermediates by reaction of aromatic compounds with nitrogen dioxide is, of course, limited to the more reactive substrates such as phenols. Hartshorn’s group24 33, also recently in conjunction with Eberson34 39, having investigated the reactions of ipso-compounds formed from many different substituted phenols using mainly fuming nitric acid as nitrating agent, wished to determine whether nitrogen dioxide would give a similar pattern of reactivity27. In a typical experiment, a suspension of 3,4,5-tribromo-2,6-dimethylphenol was suspended in cyclohexane, the solvent deoxygenated by a flow of nitrogen, and pure nitrogen dioxide was bubbled through the stirred suspension. A detailed study of the reaction has demonstrated the occurrence of the appropriate 4-nitrocyclohexa-2,5-dienone, as well as a product, 6-hydroxy-2,6-dimethyl- 3,4,5-tribromocyclohexa-2,5-dienone, which further reacted with NO2 to generate two isomeric dinitro ketones (10, cis- 56%; 11, trans- 31%). The possible sequence of reactions is illustrated in Scheme 2. Several points of interest arise here: some will have to be
21. Displacement and ipso-substitution in nitration |
955 |
||||
Br |
|
Br |
|
|
Br |
|
|
|
|
||
Br |
Me |
Br |
Me |
Br |
Me |
|
|
||||
Br |
OH |
Br |
O |
Br |
O |
|
|
||||
Me |
|
Me |
NO2 |
Me |
ONO |
Br |
|
|
Br |
|
|
Br |
|
Me |
Br |
Me |
|
|
|
|
|||
Br |
|
NO2 |
|
|
|
|
|
|
|
|
|
NO2 |
|
O |
Br |
O |
|
OH |
|
Me |
OH |
|
|
Me |
|
|
|||
|
|
SCHEME 2 |
|
|
|
|
|
Br |
|
Br |
|
Br |
|
Me |
Br |
NO2 |
|
Br |
|
NO2 |
Br |
Me |
|
|
|
|
|
||
NO2 |
|
O |
NO2 |
O |
|
Me |
OH |
OH |
|
||
|
Me |
|
|||
|
|
(10) |
|
(11) |
|
deferred to a later moment when we consider the rearrangements of ipso-intermediates. The suggested route to the formation of the ipso-intermediate, hydrogen atom abstraction from the hydroxy group of the phenol by NO2, to generate the corresponding phenoxy radical which then combines with a further molecule of NO2, is reminiscent of the radical process which occurs in nitrous acid catalysed nitration (vide supra). That this completely homolytic process, carried out in a non-polar solvent such as cyclohexane, should generate the same final products, 10 and 11, in the same ratio (10, 54%; 11, 30%) as the reaction in fuming nitric acid, is remarkable.
Confirmation |
of |
this |
mechanism, |
first |
proposed |
by |
Brunton and coworkers40, |
||||
for |
the |
generation |
of |
the |
ipso-intermediates from phenols by NO2 has |
been |
|||||
produced |
by Coombes and collaborators41,42. They |
examined the kinetics of |
|||||||||
the |
reactions |
of |
phenol, |
4-methylphenol and some 2,4,6-trialkylphenols |
with |
||||||
NO2 |
in |
cyclohexane. |
For |
example, |
the |
reaction |
of |
2,6-di-t-butyl-4-methylphenol |
|||
with NO2 |
forms |
the corresponding 2,6-di-t-butyl-4-methyl-4-nitro-cyclohexa-2,5- |
||||
dienone (12) |
with |
a |
second order dependence of rate |
on the |
concentration |
|
of NO2; |
this |
is |
in |
accordance with the mechanism |
outlined |
in Scheme 3. |
On the other hand, the same reaction of 2,4,6-trimethylphenol is independent of the concentration of NO2. This behaviour is only explicable if in the latter case the formation of the ipso-intermediate occurs by a rate limiting process from some other, rapidly formed, intermediate. In order to investigate this process further, 2,4,6-tri-t-butylphenol

956 |
J. P. B. Sandall |
|
|
Me |
But |
|
|
NO2 |
ArOH + NO2 |
|
ArO + HNO2 |
|
|||
|
|||
ArO + NO2 |
|
|
|
|
|
(12) |
|
|
|
||
|
|
||
|
O |
|
|
But
(12)
SCHEME 3
was reacted with NO2; it generated a small quantity of 2,6-di-t-butyl-p-benzoquinone (3%), the expected ipso-intermediate (67%) and a compound (22%) shown by spectroscopic data to have structure 13. This same compound can be formed in 42% yield using NO2 in carbon tetrachloride solution but not apparently in benzene28. A kinetic study on the rate of formation of these compounds, together with data for the rate of reaction of NO2 with the 2,4,6-tri-t-butylphenoxy radical43, was completely consistent with Scheme 4, with the radical lying on the reaction path. It was suggested that the intermediate in this scheme may be identified with the ipso-intermediate generated by addition of NO2 at the 2- rather than the 4-position; this intermediate could be observed at low temperatures in deuteriochloroform; it isomerized to the 4-ipso-product as the temperature was raised. In the case of the more bulky t-butyl groups, the rate of isomerization becomes relatively fast.
|
O |
|
|
|
|
|
But |
But |
ArOH + NO2 |
|
|
ArO + HNO2 |
|
|
|
|
||||
|
OH |
|
|
|||
|
|
|
||||
|
ArO + NO2 |
|
|
(12, tris-But ) + Intermediate |
||
|
H |
|
|
|||
|
Intermediate |
|
|
(13) |
||
|
|
|
||||
But |
NO2 |
|
|
|
|
|
NO |
|
|
|
|
|
|
|
2 |
|
|
|
|
|
(13)
SCHEME 4
Coombes and coworkers42 have also investigated the reaction of 4-methylphenol with NO2 in cyclohexane as solvent. The reaction at room temperature give 32% 4-methyl-4- nitrocyclohexa-2,5-dienone and 68% 4-methyl-2-nitrophenol. This may be compared with the 30% and 70% respectively reported above for the nitrous acid catalysed process in acetic acid; again a fascinating similarity. The kinetics could only be explained in terms of a second intermediate, A in Scheme 5; such intermediates have been observed previously in the nitration of 2,5-dimethylphenol. In confirmation of this, a primary kinetic isotope effect was observed in the nitration of phenol itself at a relatively low NO2 concentration; and the product ratio of ortho- to para-substitution was shown to be almost identical to that for the nitrous acid catalysed nitration mechanism. Clearly, the product determining stage for both this NO2 attack and for the nitrous acid catalysed process is the addition reaction of the phenoxy and NO2 radicals. In fact, the observed product ratio is close to that predicted by relative magnitudes of the calculated (VAMP44, V.5.01, AM1 and PM3) annihilated spin densities (32, 36, 32) of the phenoxy radical at the 2-, 4- and 6-positions.
In general, it would appear that the process of ipso-intermediate formation of the more reactive substrates such as phenols and amines is dominated by nitrous acid catalysed

21. Displacement and ipso-substitution in nitration |
957 |
||||
OH |
O • |
|
|
||
|
NO2 • |
+ HNO2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
CH3 |
|
|
||
|
|
• |
NO2 • |
|
|
|
NO2 |
|
|
|
|
|
O |
O |
|
|
|
|
|
H |
|
|
|
|
|
|
NO2 |
|
|
|
CH3 |
Me |
NO2 |
|
|
|
|
|
|
||
(A)
SCHEME 5
nitration, unless specific precautions are taken for this to be eliminated, and that this radical process also gives higher yields of the intermediates when the necessary substituent groups are in the 1,4-positions. In the case of dialkylbenzenes and halogenoalkylbenzenes, the classical Ingold mechanism appears more probable, although it is unlikely that the incipient ipso-carbocation escapes from the encounter pair in solvents such as acetic anhydride at low temperatures.
III.FURTHER REACTIONS OF ipso-INTERMEDIATES AND ADDUCTS A. Solvolysis of Adducts
In this section we will emphasize the solvolytic behaviour of the ipso-adduct, although discussion of rearrangements cannot be avoided, since the latter are concurrent, competing reactions. Again, to some extent, the behaviour of the adducts arising from the nitration of 1,4-dimethylbenzene may be used as a general example. Several groups have investigated the solvolysis of 1,4-dimethyl-4-nitrocyclohexa-2,5-dienyl acetate (1, 14) and its corresponding dienol. Myhre and collaborators45 examined both products and kinetics in aqueous ethanol as solvent, comparing product formation for these systems with that in 50% 80% aqueous sulphuric acid. The E and Z isomers of both ester and alcohol were prepared and characterized as outlined above and subjected to solvolytic elimination in aqueous ethanol (40% 75%). Both isomeric acetates generated 2,5-dimethylphenyl acetate in at least 99.5% yield; no detectable amounts of the corresponding 2,4-isomer were found. The decomposition was first order in adduct, the E isomer being more reactive (by a factor of about 3). In acid solution, the products were, depending on the acid strength, a mixture of 2,5-dimethylnitrobenzene and the side-chain substitution product, p-tolualdehyde. Thus in greater than 77% acid, the nitro compound was obtained in quantitative yield, whereas in 64% acid, while the total yield was still quantitative, it comprised 72% nitro compound and 26% aldehyde. At still lower acid concentrations, the yield of nitro compound fell off drastically, with no further increase in aldehyde production, although other side-chain

958 |
J. P. B. Sandall |
substituted compounds were observed. The kinetics in these solutions were too fast to measure by conventional techniques.
The results in aqueous ethanol were discussed in terms of Scheme 6. The rate limiting process is the rate of the solvolytic elimination of nitrous acid which depends on the ionizing power of the solvent; no primary isotope effect was observed. The migration of the acetate group had previously been shown to be intramolecular and involving a 1,2-shift46, although work with pseudocumene47 has shown the possibility of a 1,3-shift of the acetoxyl group in dilute acid. The bridged cation is suggested on the basis of the known stability of such structures. The kinetic data for the aqueous alcoholic solvolysis of the dienol are more difficult to interpret since the reaction is more complex, producing about 30% 2,4-dimethylphenol, 50% 1,4-dimethylcyclohexadiene-1,4-diol together with smaller quantities of other species. The results were however thought to be consistent with Scheme 7, where the methyl group migrates more easily than the hydroxyl group. Possible routes to the variety of products obtained in acid solution are also outlined in this work.
A thorough re-examination of the products of solvolysis in aqueous organic solvents of the acetate (14) and its corresponding dienol and methyl ether was carried out by Fischer, Henderson and Smyth9. In order to maximize the solvolysis of the nitro group, 50% aqueous methanol was used. For up to 10 half-lives for the initial solvolysis, the products from the nitro dienol consisted of mainly methoxy dienol (15) and dienediol (16) together with the two phenols 17 and 18 (Scheme 8). At very long reaction times, the initially formed dienes were aromatized forming dimethylphenols and anisoles. In the presence of base, the rearomatization was suppressed; in trifluoroacetic acid, the dienediol gave close to 100% of the phenol (17). All the products were accounted for in terms of the reactions of the substituted 1,4-dimethyl-4-nitro-2,5-cyclohexadienyl cation, which can undergo 1,2- or 1,4-nucleophilic addition generating new cyclohexadienes. It is also suggested that the cation can also undergo competing rearrangements of the methyl group with the acetyl, hydroxyl or methoxyl substituents. Finally, the cation may lose a proton from the methyl side-chain to generate a triene, which in turn will undergo side-chain substitution via a benzylic cation. The variation in product yields with reaction conditions could be rationalized on this basis.
Further light has been thrown on the mechanism of solvolysis of ipso-adducts in aqueous acid solution by kinetic studies48,49. In order that the reactions should proceed at a measurable rate, substrates with more electron withdrawing groups than alkyl had to be used. The authors examined the kinetics and products of aqueous sulphuric acid solvolyses of 2-cyano-3,4-dimethyl-4-nitro-cyclohexa-2,5-dienyl acetate (19) and 5-chloro-2-methyl- 2-nitrocyclohexa-3,5-dienyl acetate (20, a 1,2-adduct). When 20 was heated in acetic acid50, 2,3-dimethylbenzonitrile and 2,3-dimethyl-5-nitrobenzonitrile were formed and it was suggested that the nitro group migration was, somewhat unusually, an intramolecular 1,3-migration more often these are 1,2-shifts. Moodie and coworkers48,49 therefore investigated both kinetics and products in aqueous acid solution in the hope of clarifying these points. Their general mechanism is set out in Scheme 9, where the primary process is considered to be a competition between the elimination of nitrous acid (E1) to generate the Wheland type intermediate (21) that is the precursor of the aromatic ester, and the acid catalysed hydrolysis of the acetate group (AAl1) to form the ipso-cation (22). The cation underwent extensive capture by water particularly at low acidities and a 1,2-rearrangement of the nitro group, rather than the 1,3-shift, observed on heating. These rearrangements will be discussed later. A very similar picture was obtained for the ipso-adduct 20: competing acid-catalysed elimination of nitrous acid and ester solvolysis. The ipso-cation generated by this solvolysis is identical to that obtained by attack on 4-chloromethylbenzene; no less than 59% of primary attack by nitronium ion takes place at C-1 in this compound.