

Supplement A3: The Chemistry of Double-Bonded Functional Groups. Edited by Saul Patai Copyright 1997 John Wiley & Sons, Ltd.
ISBN: 0-471-95956-1
CHAPTER 22
Radical anions and radical cations derived from compounds containing C=C, C=O or C=N groups
DANIEL J. BERGER and JAMES M. TANKO
Department of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061-0212, USA
Fax: 540-231-3255 e-mail: JTANKO@VT.EDU
I. ABBREVIATIONS . . . . . . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1282 |
||
II. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1283 |
||
III. RADICAL IONS OF >CDO CONTAINING COMPOUNDS . . . . . . . |
1283 |
|||
A. >CDO Radical Cations . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1283 |
||
B. >CDO Radical Anions . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1283 |
||
1. Methods of generation . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1283 |
||
|
|
D |
|
|
2. Reactions of >C Ož . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1285 |
||
a. Free ions, ion pairs or neutral free radicals? |
. . . . . . . . . . . . . |
1285 |
||
b. Overview . . . . . . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1286 |
||
|
|
D |
|
|
c. Reactions of >C Ož with radicals or radical ions |
|
|||
(pinacolization) . . . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1288 |
||
|
|
D |
|
|
d. Fragmentations of >C Ož . . . . . . . . . . |
. . . . . . . . . . . . . |
1289 |
||
(i) |
˛- or ˇ-cleavage of carbonyl radical anions . . . . . . . . . . |
1289 |
||
|
|
D |
|
|
(ii) |
Long-range cleavage involving >C Ož . . . . . . . . . . . |
1292 |
||
|
|
D |
|
|
(iii) |
Ring-opening reactions involving >C |
Ož . . . . . . . . . . |
1293 |
|
|
(a) |
Cyclopropane ring openings . . . . |
. . . . . . . . . . . . . |
1293 |
|
(b) |
Oxirane ring openings . . . . . . . . |
. . . . . . . . . . . . . |
1299 |
|
(c) |
Aziridine ring openings . . . . . . . |
. . . . . . . . . . . . . |
1302 |
|
(d) Fourand five-membered ring openings . . . . . . . . . . |
1304 |
||
|
|
D |
|
|
e. Addition of >C Ož to -systems (ketone |
olefin coupling |
|
||
reactions) . . . . . . . . . . . . . . . . . . . . . . . |
. . . . . . . . . . . . . |
1305 |
||
(i) |
Intermolecular additions . . . . . . . . . . |
. . . . . . . . . . . . . |
1305 |
|
(ii) |
Intramolecular additions . . . . . . . . . . |
. . . . . . . . . . . . . |
1307 |
|
1281
1282 |
|
Daniel J. Berger and James M. Tanko |
|
|
||||||
|
|
(a) Intramolecular additions to remote alkene or allene |
|
|
||||||
|
|
functionalities . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1307 |
|
|
|
(b) Intramolecular additions to other remote -systems . . |
1312 |
|
||||||
|
f. Radical anions generated from ˛,ˇ-unsaturated carbonyl |
|
|
|||||||
|
compounds . . . . . . . . . . . . . . . |
.D. |
. . . .D. . . . . . . . . . . . . . |
1315 |
|
|||||
|
(i) |
|
1315 |
|
||||||
|
‘Nucleophilic’ reactions of >C |
|
C C Ož . . . . . . . . . |
|
||||||
|
(ii) |
D |
|
C |
D |
Ož |
1317 |
|
||
|
‘Radical’ reactions of >C C |
|
|
|
||||||
|
(iii) |
Miscellaneous ketone (aldehyde)/activated olefin |
|
|
||||||
|
|
couplings . . . . . . . . . . . . . |
. |
. |
. |
. . . . |
. |
. . . . . . . . . . . |
1317 |
|
IV. RADICAL IONS OF >CDC< CONTAINING COMPOUNDS . . . . . . |
1318 |
|
||||||||
A. Chemistry of Radical Cations Derived from Carbon |
|
Carbon |
|
|
||||||
|
|
|
||||||||
Double Bonds . . . . . . . . . . . . . . . . . |
. |
. |
. |
. . . . . |
|
. . . . . . . . . . . . |
1318 |
|
||
1. |
Methods of generation . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1319 |
|
|
|
|
D |
|
|
|
|
|
|
1319 |
|
2. Reactions involving >C C<žC . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
|
|||
|
a. Cation radical ‘pericyclic’ reactions |
|
. . |
. . . . |
. |
. . . . . . . . . . . |
1319 |
|
||
|
(i) |
The ‘hole-catalyzed Diels-Alder’ reaction . . . . . . . . . . . |
1319 |
|
||||||
|
(ii) |
Vinylcyclopropane rearrangements |
. . . |
. |
. . . . . . . . . . . . |
1323 |
||||
|
(iii) |
Epoxidations of alkenes . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1327 |
|
|
(iv) |
Cyclopropanations of alkenes |
|
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1331 |
|
|
b. Oxidation/reduction of alkenes . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1331 |
|
|
|
(i) |
Hydrogenation . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1331 |
|
|
(ii) |
Oxygenation . . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1331 |
|
|
c. Nucleophilic addition to alkenes promoted by one-electron |
|
|
|||||||
|
oxidation . . . . . . . . . . . . . . . . |
. |
. |
. . |
. . . . |
|
. . . . . . . . . . . . |
1332 |
|
|
|
d. Oxidative C C bond-forming reactions |
. . . . |
. |
. . . . . . . . . . . |
1333 |
|
||||
|
|
D |
|
|
|
|
|
|
1338 |
|
3. Reactions involving >C C<žC . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
|
|||
|
a. Z ! E isomerization . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1340 |
|
|
|
b. Reduction/dimerization of alkenes |
|
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1340 |
|
|
|
c. The vinylic SRN 1 reaction . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1341 |
|
|
V. RADICAL IONS OF >CDN CONTAINING COMPOUNDS . . . . . . |
1343 |
|
||||||||
A. >CDN Radical Cations . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1343 |
|
||
1. Overview . . . . . . . . . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1343 |
|
||
2. Reactions of >CDN žC . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1343 |
|
||
B. >CDN Radical Anions . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1345 |
|
||
VI. CLOSING REMARKS . . . . . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1348 |
|
||
VII. ACKNOWLEDGMENT . . . . . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1348 |
|
||
VIII. NOTES AND REFERENCES . . . . . . . . |
. |
. |
. . |
. . . . |
. |
. . . . . . . . . . . |
1348 |
|
||
|
|
|
|
|
|
|
|
|||
|
|
I. ABBREVIATIONS |
|
|
|
|
|
|||
BSA |
benzeneseleninic anhydride |
|
|
|
|
|
|
|
|
|
CRIP |
contact radical ion pair |
|
|
|
|
|
|
|
|
|
DCA |
dicyanoanthracene |
|
|
|
|
|
|
|
|
|
DCB |
dicyanobenzene |
|
|
|
|
|
|
|
|
|
DCN |
dicyanonaphthalene |
|
|
|
|
|
|
|
|
|
DMPC |
dimethylpyrrolidinium ion |
|
|
|
|
|
|
|
|
|
FI |
free ion |
|
|
|
|
|
|
|
|
|
MCPBA m-chloroperbenzoic acid |
|
|
|
|
|
|
|
|
||
PIET |
photo-induced electron transfer |
|
|
|
|
|
|
|
|
|
SET |
single electron transfer |
|
|
|
|
|
|
|
|
|
SSRIP |
solvent-separated radical ion pair |
|
|
|
|
|
|
|
|
|
TIET |
thermally induced electron transfer |
|
|
|
|
|
|
|
|
|
TPP |
triphenylpyrilium |
|
|
|
|
|
|
|
|
|
22. Radical anions and cations derived from CDC, CDO or CDN groups 1283
II. INTRODUCTION
The addition or removal of an electron from a closed-shell organic molecule is a fundamental mechanism for increasing chemical reactivity. Enhanced reactivity results from the diminution of bond orders in the molecule (as a consequence of removing electrons from bonding orbitals or adding electrons to anti-bonding orbitals) and the formation of a charged, paramagnetic species (i.e. radical cations or radical anions). The focus of this chapter is on the chemistry of radical ions generated from >CDC<, >CDN- and >CDO functionalities, with a particular emphasis on reactions which either have synthetic utility (or the potential thereof), or which are interesting from a mechanistic perspective. The review covers the period 1985 1995.
III.RADICAL IONS OF >C=O CONTAINING COMPOUNDS A. >C=O Radical Cations
In the gas phase, the chemistry of radical cations generated from >CDO con-
taining species is well-documented, especially |
in the field of mass spectrometry |
|||||||
where a number of diverse and particularly |
diagnostic |
processes are |
known such |
|||||
as ˛-cleavage (R(CO)RžC ! RCOC C Rž) |
and |
the |
McLafferty |
rearrangement |
||||
(R COžC CH2CH2CH3 |
! |
R(COH)CHžC |
C |
CH2 |
D |
CH2 1. In solution, however, |
||
|
2 |
|
|
|
|
|||
reports of radical cations generated from >CDO containing organic molecules are rare. (For the oxidation of ketones, it is generally the enol form which is actually oxidized)2
B. >C=O Radical Anions
1. Methods of generation
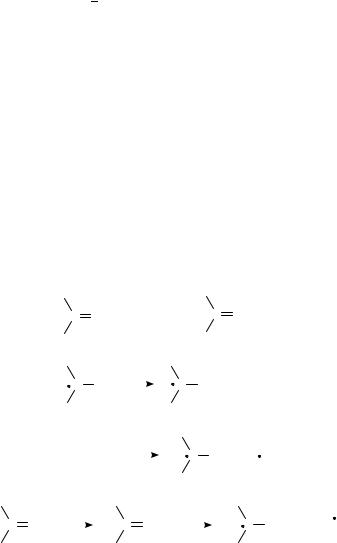
Radical anions generated from ketones (or aldehydes) are referred to as ketyl anions. Common methods for generation of >CDOž are summarized in Figure 1.
(1) Direct chemical or electrochemical reduction of carbonyl compounds
C O + e− 
 C O−
C O−
(2) |
Deprotonation of ketyl radicals |
|
|
|
|
|
||||||||||
|
|
|
|
C |
|
OH |
|
|
|
C |
O− + |
H+ |
||||
|
|
|
|
|
||||||||||||
(3) |
α-Hydrogen abstraction from alkoxides |
|
|
|
||||||||||||
|
H |
|
|
|
|
O− |
|
|
C |
O− |
+ H |
|||||
|
|
|
|
|
|
|
|
|||||||||
|
|
C |
|
|||||||||||||
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
(4) |
Photo-induced electron transfer (PIET) |
|
|
|
||||||||||||
|
C O |
|
hν |
|
|
C O* |
|
R3 N |
C O− + R3 N + |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
FIGURE 1. Generation of >CDOž in solution
1284 |
Daniel J. Berger and James M. Tanko |
The simplest method for generation of a ketyl anion is via direct reduction (chemically of electrochemically) of an aldehyde or ketone (Figure 1, reaction 1). Historically, it was via direct reduction of a ketone with an alkali metal that ketyl anions were first discovered.
In 1891, Beckman reported that reaction of benzophenone with sodium resulted in a deep blue colored solution3. Schlenk and Weickel later suggested that Ph2CDOž NaC was the species responsible for the blue color4. (Ph2CDOž MC generated in this manner is a common laboratory reagent for the purification of ether solvents.)
Ketyl anions can also be formed via the abstraction of a proton from a neutral ketyl radical (Figure 1, reaction 2). This process is best depicted as an equilibrium (vide infra)5.
Although less common, ketyl anions can also be generated by removal of an ˛-hydrogen from an alkoxide (Figure 1, reaction 3). An interesting example where a ketyl anion is formed as an intermediate in this manner is provided by the electrochemically-initiated reduction of an aryl halide by an alkoxide anion via the free radical chain process illustrated in Scheme 16.
|
|
|
ArX |
|
Ar |
+ |
X− |
O− |
|||
|
|
|
|
||||||||
Ar |
+ |
CH |
O− |
|
|
ArH |
+ |
C |
|||
|
|
||||||||||
•C |
|
O− |
+ ArX |
|
C |
O + ArX |
|
|
|||
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
ArX |
+ |
CH |
O− |
|
ArH |
+ |
C |
O |
+ X− (overall) |
||
SCHEME 1
Ketyl anions can be generated photochemically via PIET (Figure 1, reaction 4). In this method, the carbonyl is excited photochemically to its triplet state which is readily reduced by a 3° amine7. Initially a ketyl anion/ammonium radical cation pair (>CDOž /R3NžC ) is produced. Several distinct ion-pair intermediates have been characterized: contact radical ion pairs (CRIPs), solvent-separated radical ion pairs (SSRIPs) and free ions (FIs). Because each of these may produce different products (e.g. CRIPs may lead to reactions between >CDOž and R3NžC whereas unimolecular processes may be favored by the free ions), control over the efficiency of CRIP, SSRIP and FI production may be an important consideration in order to maximize the yield of the desired product8. Sideproducts may also arise as a result of the follow-up chemistry of R3NžC . In addition to possessing radical and cationic character, R3NžC is also an acidic species (pKa ³ 10 for (p-MeOC6H4)2NCH3žC 9 and thus subject to deprotonation (equation 1). (In fact, >CDOž may itself serve as the base producing a neutral ketyl radical, >C(ž)OH.)
+ |
C NR2 + H+ |
(1) |
H C NR2 |
Finally, radical anions of carbonyl-containing compounds are often produced ‘unexpectedly’ in reactions involving nucleophiles or easily oxidized free radicals. In a seminal 1964 report, Russell, Janzen and Strom noted that ketyl anions could be detected by ESR
22. Radical anions and cations derived from CDC, CDO or CDN groups 1285
when neutral ketones were treated with a number of common nucleophilic reagents10. Since that time, the question as to whether a number of simple organic transformations involving nucleophilic addition to carbonyl compounds proceeds via the traditional polar (two-electron) pathway or via single electron transfer (SET), paths a and b, respectively (Scheme 2), has attracted a great deal of attention. A detailed analysis of the claims (and counterclaims) of electron transfer in nucleophilic and/or radical additions to >CDO is beyond the intended scope of this chapter. It suffices to say that during the period covered by this review, the role of electron transfer has been extensively examined and debated in a large number of reactions involving aldehydes, ketones and other carbonylcontaining compounds (e.g. the Grignard reaction11, Clemmenson reduction12, Aldol condensation13, Wittig reaction14, Meerwein Pondorff Verley reduction15, reactions with RLi16, R2NLi17, NADH analogues18, complex metal hydrides19 and radical-mediated reductions involving R3Snž20 and R3Siž)21.
O |
|
|
O− |
|
Nu:− |
(a) |
|
||
+ |
C |
|||
polar |
||||
C |
|
|
||
|
|
|
Nu |
|
SET |
|
|
|
|
(b) |
|
O− |
|
|
|
|
|
||
Nu |
+ |
C |
|
SCHEME 2
2.Reactions of >CDOž
a. Free ions, ion pairs or neutral free radicals? Regardless of how it is produced, charge balance requires that >CDOž be generated in solution with an appropriate counterion, which in most cases will be a metal cation (MCn). In general, the precise effect of the counterion on reactivity is not very well understood. In principle, a continuum of possible intermediates may exist in solution ranging from free (unassociated) ions $ ion pairs $
covalent adduct (Scheme 3)22.
C O− + M+ |
|
C O−Μ+ |
|
C O M |
|
|
|||
|
|
|||
free ions |
|
ion pair |
|
covalent |
|
|
adduct |
||
|
|
|
|
|
|
|
SCHEME 3 |
|
|
Early ESR studies demonstrated that the hyperfine coupling constant (ac13 ) for 13C(car- bonyl)-substituted fluorenone radical anion is counterion-dependent. For the ‘free’ ion, ac13 D 2.75 Gauss. In contrast, when the counterion is LiC, ac13 D 6.2 Gauss23. Consider Scheme 4: For the ‘free’ ion, canonical structure 1 and 2 are contributors to the resonance hybrid. For the >CDOž / LiC ion pair, association of LiC with oxygen increases the relative contribution of canonical structure 1 to the resonance hybrid, resulting in greater spin density at carbon. The fact that spin (and charge density) varies as a function of counterion (and presumably solvent) will certainly affect the reactivity of the radical ion. However, very few quantitative studies exist which directly address this point.
1286 |
Daniel J. Berger and James M. Tanko |
|||
|
O− |
|
|
O |
|
C |
|
|
C |
|
|
|
||
|
|
|
|
− |
|
(1) |
(2) |
||
|
|
SCHEME 4 |
||
It is likely that many of the reactive intermediates discussed in this chapter are nearing the tight ion pair ! covalent adduct end of this continuum (e.g. reactions of >CDO with SmC2, R3Snž, etc.). Our operational criterion for inclusion of a reaction as involving a radical anion centers on whether the paramagnetic intermediate involved in the transformation is sufficiently anionic so as to have nucleophilic properties (i.e. can it be alkylated? protonated?).
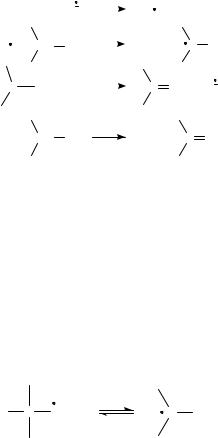
b. Overview. Figure 2 summarizes most of the significant reactions of radical anions generated from carbonyl compounds.
The most fundamental reaction of >CDOž involves electron transfer to another substrate (Figure 2, reaction 1). While at first this process might seem trivial, carbonyl compounds have proven effective electron transfer mediators for the reduction of a variety of substrates.
This mediated reduction process has been utilized extensively in electrochemistry on both a preparative and analytical scale, and is often catalytic. In this process, a mediator, whose reduction potential is more positive than that of the substrate, is reduced electrochemically. The resulting radical anion subsequently transfers an electron to the substrate, regenerating the mediator. The radical anion of the substrate then undergoes further chemistry. Use of a carbonyl compound as a mediator is not mandatory. Carbonyl compounds offer the advantage that with appropriate substitution, their reduction potentials can be tailored over a wide range to suit a large variety of substrates and reactions. This technique is highly effective for obtaining important kinetic and thermodynamic information for substrates which cannot be studied by direct electrochemical methods (e.g. cyclic or
linear sweep voltammetry). The interested reader is directed to the work of Saveant´24 and Lund25.
Another fundamental reaction of >CDOž involves its reactivity as a base. In the Brønsted sense, >CDOž may react with a proton donor to produce a neutral ketyl radical (>C(ž)OH, Figure 2, reaction 2). This is an important process when the reduction of a carbonyl compound is carried out under acidic conditions or in a protic media (e.g. electrochemically, with less reactive reducing reagents such as Mg or Zn, or when >CDOž is produced via PIET and R3NžC has available ˛-protons). The follow-up chemistry of >C(ž)OH is that of a neutral free radical (dimerization to form pinacols, addition to unsaturated compounds, fragmentations/ring-openings, etc.), and thus beyond the scope of this chapter.
Reaction 3, Figure 2, illustrates another variant, the reactivity of >CDOž as a Lewis base (e.g. nucleophile) via reaction with electrophilic species. In the specific case of reacting with an alkyl halide (R X), a direct nucleophilic displacement (SN2) process was initially envisioned. However, it is now evident that these alkylations take
place via initial SET: CDOž C R X ! CDO C Rž C X , followed by Rž C CDOž ! R C O . The interested reader is directed to the work of Garst and coworkers26. In addition, there has been considerable interest in this chemistry from the perspective of the dynamics and theory of dissociative electron transfer, and the interested reader is directed to the work of Saveant´27 and Lund.28 (Evidence has recently been presented for a continuous SN2/SET mechanistic spectrum for the intramolecular reactions of ketyl anions and alkyl halides, Scheme 5)29.
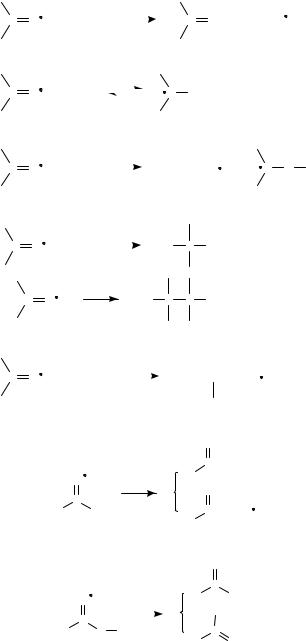
22. Radical anions and cations derived from CDC, CDO or CDN groups 1287
(1) |
electron transfer |
|
|
|
|
|
|
|
|
|
||||
|
C O − + substrate |
|
|
|
|
C |
O |
+ |
substrate − |
|||||
|
|
|
|
|
||||||||||
(2) |
proton transfer |
|
|
|
|
|
|
|
|
|
||||
|
C O − + HA |
|
|
|
|
C OH |
+ |
A− |
||||||
|
|
|
|
|||||||||||
|
|
|
|
|||||||||||
(3) |
reaction with electrophiles |
|
|
|
|
|
|
|
|
|
||||
|
C O − + E+ |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
E |
|
C |
|
O or |
C O E |
|||||||
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(4) reaction with radicals or radical ions
C O − + R
 R C O−
R C O−
|
or |
2 C O − |
−O C C O− |
(5) addition to π-systems
C O − + X |
|
Y |
|
−O |
|
|
|
|
|
|
|
C |
|
X |
|
Y |
|||||||
|
|
|
|
||||||||
|
|
|
|
|
|||||||
|
|
|
|
||||||||
(6) fragmentation
O
(a) α-cleavage
C + Z−
+ Z−
|
O − |
R |
|
O |
|
|
C |
|
|
C− + Z |
|
R |
Z |
|
|
|
R |
(b) β-cleavage
|
|
|
O |
|
|
|
|
|
|
C |
X• |
+ |
Y− |
|
O − |
|
R |
|
|
|
|
|
O− |
|
|
|
|
|
C |
|
|
|
|
|
|
|
|
|
+ |
Y• |
|
R |
X Y |
|
C |
X |
||
|
|
|
R |
|
|
FIGURE 2. Reactions of >CDOž
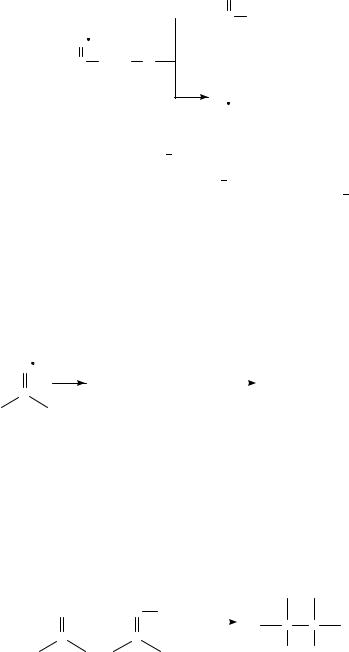
1288 |
Daniel J. Berger and James M. Tanko |
O
 ArC (CH2 )n
ArC (CH2 )n + X−
+ X−
SET
O −
ArC (CH2 )n X
O |
|
|
CH2 |
+ X− |
|||
|
|
||||||
ArC |
|
|
|
|
|
|
|
|
|
|
(CH2 )n−1 |
|
|||
|
|
|
|
||||
SN2
SCHEME 5
With regard to Figure 2, reactions 1 3: Because the recent work dealing with these reactions is only tangentially related to ketyl anions, these reactions will not be considered further in this chapter. The remaining reactions (4 6) depicted in Figure 2 represent the bulk of the mechanistic and synthetic work which was reported in the 1985 1995 period, and will be the primary focus of the remainder of this section.
c. Reactions of >CDOž with radicals or radical ions (pinacolization). Being paramagnetic, >CDOž is especially reactive towards free radicals and radical ions. The rate constant for reaction of LiC/benzophenonež with a 1° alkyl radical is slightly below diffusion-controlled (on the order of 1.5 ð 108 M 1 s 1 26. A similar rate constant was reported for reaction of 1° radicals with Ph(CDOž )t-Bu30. The most common reaction of >CDOž with another radical ion is dimerization to form pinacolates, which after workup yield pinacols (equation 2). The rate constant for dimerization of PhCHOž is 2.4 ð 103 M 1 s 131 .
|
O − |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
2 |
−O |
|
C |
|
C |
|
O− |
|
HO |
|
C |
|
C |
|
OH (2) |
||||||
|
|
|
|
||||||||||||||||||
|
|
|
|
||||||||||||||||||
|
C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The reduction of carbonyl compounds to form pinacol dimers can be accomplished photochemically, electrochemically or with chemical reducing agents. When conducted under acidic conditions or in protic solvents, pinacols are likely produced by coupling of two neutral ketyl radicals (vs radical anions). The electrochemical reduction is especially complicated in terms of the role of the electrode surface, counterion and solvent, and an excellent review has appeared on the subject32.
Shono and coworkers33 have recently reported a novel ketone/imine coupling process (equation 3) analogous to pinacolization. The coupling process was achieved electrochemically, utilizing a Sn cathode. As the results summarized in Scheme 6 illustrate, yields are reasonable for both interand intramolecular coupling, although diasteroselectivity was higher for the latter. The suggested mechanism for this reaction is summarized in Scheme 7.
O |
N X |
e− |
+ |
|
i-PrOH |
C |
C |
Et4 NOTs |
X = OCH3 , N(CH3 )2
C C
(3)
HO NHX
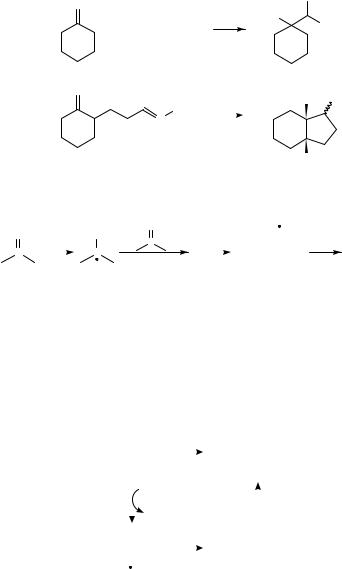
22. Radical anions and cations derived from CDC, CDO or CDN groups 1289
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
HO |
|
NHOCH3 |
|
|
|
|
|
|
|
|
|||||
|
|
CH3 CH |
|
NOCH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
93% |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
NHOCH3 |
|
|
|
|
|
|||||
|
|
|
|
|
|
OCH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
H |
70% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
trans :cis 95:5 |
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
SCHEME 6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
O |
|
|
O− |
NOCH3 |
|
|
|
|
|
HO |
|
|
NOCH3 |
|
|
HO |
|
NHOCH3 |
|||||||||||
|
|
C |
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
e |
− |
H |
+ |
|
|
|
|
|
|
|
|
|
+e |
− |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
C |
|
|
C |
|
|
|
|
|
|
|
|
|
C |
|
C |
|
+H |
+ |
|
|
C |
|
C |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SCHEME 7
Recently, Sml2 has emerged as a potent one-electron reducing agent for carbonyl and other functional groups34. (For SmC3 C e ! SmC2, E° D 1.55 V.) As will be seen in several examples in this section, this reagent provides convenient entry into ketyl anion chemistry.
In 1983, Kagan35 reported that benzaldehyde is conveniently converted into its pinacol dimer by reactions with Sml2 in 95% yield (dl/meso 54: 44, Scheme 8). Ten years later, Kagan also reported that pinacolization of a variety of aldehydes and ketones could also be achieved with SmBr2 in virtually quantitative yield36.
O |
|
a. Sml2 |
HO |
|
|
|
|
|
|
OH |
||||
|
|
|
|
|
|
|
|
|
|
|
|
95% |
||
PhCH |
|
b. H + |
PhCH |
|
|
|
CHPh |
|||||||
|
|
|
|
|||||||||||
|
|
Sm +2 |
|
|
|
|
|
|
H + |
|||||
|
|
|
|
|
|
|
|
|||||||
|
|
Sm+3 |
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
O− |
|
|
|
|
O− |
|
|
O− |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
PhCH |
|
|
|
PhCH |
|
|
|
CHPh |
||||||
|
|
|
|
|
|
|||||||||
SCHEME 8
An intramolecular variant of this reaction was reported in 1988 by Molander and Kenny (Table 1)37. These reactions proceeded in reasonable yields and exhibited extremely high diasteroselectivity (200:1). It was suggested that the aldehyde functionality is first reduced, allowing chelation control of the stereochemistry (e.g. 3).
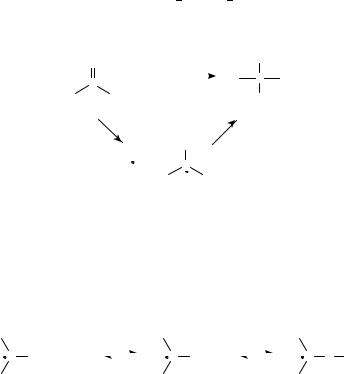
d. Fragmentations of >CDOž (i) ˛- or ˇ-cleavage of carbonyl radical anions. Radical anions derived from carbonyl compounds may undergo ˛- or ˇ-cleavage (Figure 2:
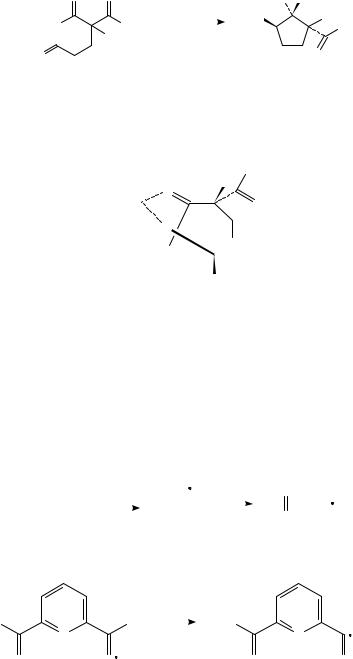
1290 |
|
Daniel J. Berger and James M. Tanko |
|
|||||
|
TABLE 1. Intramolecular pinacol coupling Sml2a |
|
||||||
|
O |
O |
|
|
|
|
R |
OH |
|
R |
|
Y |
Sml2 |
|
HO |
R′ |
|
|
R′ |
THF (MeOH) |
|
|
Y |
|||
|
|
|
|
|
|
|||
|
|
|
− 78 |
°C |
|
|
|
|
|
|
|
|
|
|
O |
||
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Y |
|
R |
|
R0 |
|
% Yield |
|
|
OCH2CH3 |
|
CH3 |
|
CH3 |
77 |
|
|
|
OCH2CH3 |
|
C6H5 |
|
CH3 |
66 |
|
|
|
N(CH2CH3)2 |
|
CH3 |
|
H |
50 |
|
|
|
a Reference 37. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Y |
|
|
|
|
|
O |
|
R′ |
|
|
|
|
|
Sm+3 |
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
− O
R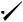
H
(3)
reactions 6a and 6b, respectively). Of the two, ˇ-cleavage is far more common. Two fragments result from this process, R(CDO)X and Y, one of which is a free radical, the other, an anion. As might be expected, the anionic fragment is generally the one which is more able to support a negative charge (i.e. the more stable anion is produced). When X and Y are incorporated in a ring, fragmentation leads to ring opening. Because of the wealth of mechanistic and synthetic interest in ring-opening reactions, this subset of ˇ-cleavage reactions is treated in a separate section.
Esters undergo ˇ-cleavage to yield carboxylate anions and alkyl radicals in accordance with equation 438. Rate constants for these fragmentations have recently been measured via pulse radiolysis, and it was found that the rate of the reaction increases with the stability of the alkyl radical39.
O |
O − |
|
O |
|
(4) |
|||||
|
|
|
e− |
|
|
|
|
PhCO− |
+ R |
|
|
|
|
|
|
|
|
||||
PhCOR |
PhCOR |
|
|
|
||||||
Thioesters, on the other hand, appear to undergo ˛-fragmentation to yield acyl radicals and thiolates (equation 5)40.
C3 H5S |
N |
SC3 H5 |
|
C3 H5S |
N |
+ C3 H5S - |
|
||||||
|
|
|
|
|
||
O |
O − |
|
|
O |
O |
(5) |
|
|
|
|
|
|