

12. The electrochemistry of the CDC, CDO and CDN groups |
631 |
B. Anodic Oxidation
1. Aldehydes and ketones
Direct electrochemical oxidation of ketones and even aldehydes is difficult to achieve, paralleling their chemical resistance to oxidation. A number of reports have however appeared in which chemical transformation of ketones is effected by anodically generated positive halogen species. Previous work in this area has been reviewed89. Typically, oxidation of a halide ion produces a halogenating agent (the exact chemical structure of which is often uncertain) which reacts with the carbonyl compound to afford an alpha- halo derivative. Usually the latter undergoes a follow-up reaction; a surprising variety of the latter have been observed, depending on experimental conditions, the identity of the halide ion and the structure of the carbonyl compound. Recent examples of this type of chemistry include the anodic conversion of methyl ketones to methyl esters in methanol containing sodium bromide (an electrochemical haloform reaction)90, the oxidative conversion of ketones into epoxy nitriles (66) in the presence of sodium cyanide and potassium iodide (equation 33)91 and the oxidative rearrangement of arylalkyl ketones (67) to esters (68) in the presence of iodine and trimethyl orthoformate92. The mechanism shown in Scheme 16 for the conversion of 67 to 68 appears simpler than that proposed by the group of Shono92.
|
|
|
R2 |
|
|
|
|
|
|
|
[O] |
|
|
|
|
|
O |
|
|
||
|
|
|
|
|
|
R3 |
|
|
|
|
R2 |
R3 |
|
||||||||
|
|
R1 |
NaCN/MeOH |
(33) |
|||||||||||||||||
|
|
|
|
|
|
|
|
R |
1 |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
KI (cat.) |
|
|
|
|
CN |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
O |
|
|
|
|
|
|
|
(66) |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
I2 − 2e− |
|
|
2 ‘I +’ |
|
|
OMe |
|
|
|
|
|
|
Ar |
H |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
HC(OMe)3 |
|
|
|
|
|
|
|
|
− MeOH |
|
|
|
|||||||
ArCOCH2 R |
|
|
ArCCH R |
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
MeO |
R |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
OMe |
|
|
|
|
|
|
|||||
(67) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ar |
H |
|
|
|
|
|
|
Ar |
+ |
H |
|
|
|
MeO |
H |
||||||
|
|
|
+ ‘I + ’ |
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
I |
|
|
MeOH |
Ar |
I |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
MeO |
R |
|
|
|
|
|
MeO |
|
|
R |
|
|
|
MeO |
R |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
MeO |
H |
|
|
|
|
|
|
O |
|
|
H |
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
Ar |
|
I |
|
|
|
|
|
|
MeO |
|
|
Ar |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
MeO |
R |
|
|
|
|
|
|
|
R |
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
(68) |
|
|
|
|
|
|
|
|
|
SCHEME 16
One of the few anodic transformations of ketones in the presence of halogen derivatives which appears to involve anodic oxidation of the carbonyl compound itself is shown in
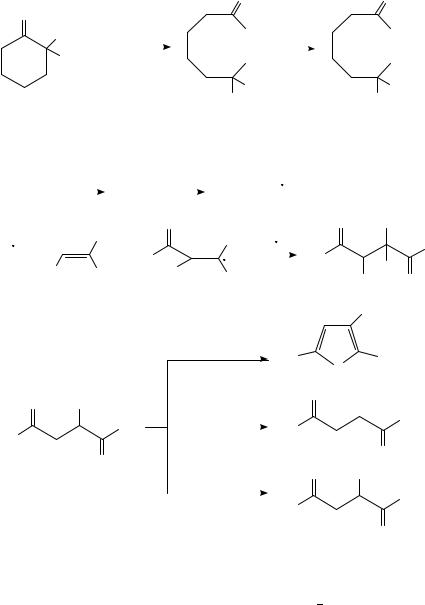
632 |
Albert J. Fry |
equation 3493. The stability of the uncommon tertiary fluoride functionality in methanol is noteworthy.
|
|
O |
O |
||
O |
|
|
|
|
|
R |
[O] |
F |
OMe |
||
|
|
MeOH |
(34) |
||
|
|
|
|||
R |
Et3 N . 5HF, O °C |
||||
F |
F |
||||
|
|||||
|
|
R |
R |
||
|
|
R |
R |
||
Anodic oxidation of a mixture of an aldehyde and an activated ester in aqueous acetonitrile containing lithium nitrate results in addition of two acyl groups derived from the aldehyde across the CDC double bond. These diacylated substances exhibit a diverse chemistry (Scheme 17)94.
NO3− |
− e− |
|
NO3. |
R1CHO |
|
HNO3 + R1CO |
|
|
|
||||
|
O |
|
O |
CO2 Me |
|||||||||
|
|
R3 |
|
|
|
|
|
||||||
|
|
|
|
|
R3 |
1 |
|
|
R1 |
||||
R1CO |
+ |
|
R1 |
|
|
|
|
|
R CO |
R1 |
|
|
|
|
R2 |
|
|
|
|
|
R3 |
|
|||||
|
R2 |
CO Me |
|
CO2 Me |
|
|
|
O |
|||||
|
|
2 |
|
|
|
|
|
|
R2 |
||||
|
|
|
|
|
|
|
|
|
|
|
|
CO2 Me |
|
|
|
|
|
|
|
5N H2 SO4 |
|
|
|
|
|
||
|
|
|
|
|
|
reflux, 1 h |
|
i-Pr |
|
i-Pr |
|
||
|
|
|
|
|
|
83 % |
|
|
O |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
|
O |
CO2 Me |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
2% aq NaOH |
|
|
|
|
i-Pr |
|||||
|
|
|
i-Pr |
|
|
r.t., 2 h |
|
i-Pr |
|
|
|||
i-Pr |
|
|
|
|
95 % |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
O |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
a. K2 CO3 /PhCH2 Br |
|
|
O |
CH2 Ph |
|
|||
|
|
|
|
|
b. 2 % aq NaOH |
|
|
|
|
|
|||
|
|
|
|
|
|
92 % |
|
|
i-Pr |
|
|
i-Pr |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O
SCHEME 17
2. Other carbonyl compounds
The high electropositivity of silicon means that the carbon silicon bond is readily oxidized. Yoshida has carried out an extensive study of the anodic oxidation of benzyl and allyl silanes95. Oxidation converts the silane to a carbocation, which then reacts with a nucleophilic component of the medium (Scheme 18). Yoshida has shown that the reaction
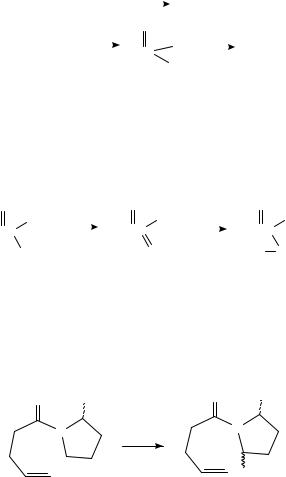
12. The electrochemistry of the CDC, CDO and CDN groups |
633 |
e
ArCH2SiMe3 ! NuSiMe3 C ArCH2ž
Nu:
ArCH2ž e ! ArCH2C ! ArCH2Nu
SCHEME 18
can be extended to the anodic oxidation of acyl silanes in the presence of a variety of nucleophiles (equation 35)96 A somewhat related study involves the oxidation of thioesters (69) in an alcohol solvent containing an iodide salt97. Replacement of sulfur by the alcohol takes place, perhaps via an activated iodosulfonium species (70) (Scheme 19).
|
|
|
|
|
|
[O] |
|
|
|
|
|
|
|
|
|
|
RCOSiMe3 |
! |
RCONu |
35 |
|||||||||
|
|
|
Nu: |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
I − − e- |
|
|
|
|
|
‘I + ’ |
O |
||||
|
|
|
|
|
||||||||||
O |
|
O |
|
|
|
|
|
|||||||
|
|
CSEt + ‘I + ’ |
|
|
RCS+ |
|
|
I |
|
R2 OH |
|
|
|
|
R1 |
|
|
|
|
|
|
|
R1 |
|
COR2 |
||||
|
|
|
|
|
|
|
Et |
|
|
|
|
|
||
(69) |
|
|
(70) |
|
|
|
|
|
|
|
|
|||
SCHEME 19
Shono and others have shown that electrochemical oxidation of amides in nucleophilic media results in incorporation of the nucleophile into the alpha-position of the N-alkyl group via an intermediate iminium species (equation 36). This so-called ‘anodic alpha- functionalization’ reaction has been shown to be of considerable synthetic value. The reaction has been extensively reviewed98.
O |
|
|
|
O |
|
O |
|
R2 |
− 2e |
− |
R2 |
Nu: |
R2 |
(36) |
|
R1CN |
|
R1CN + |
R1CN + |
||||
− H |
+ |
|
|||||
|
|||||||
|
|
|
|
|
|
|
|
CH2 R3 |
|
|
|
CHR3 |
|
Nu CHR3 |
|
Moeller has more recently contributed a series of reports describing the further use of this reaction in synthesis. Oxidation of the unsaturated amide 71 in methanolic acetonitrile afforded a substance 72 (equation 37) which could subsequently be converted into conformationally restricted peptide mimics99. Further, anodic oxidation of 73 resulted in intramolecular cyclization to afford 74 (equation 38), which could be converted into bicyclic reverse-turn peptidomimetics100. A similar intramolecular anodic cyclization of 75 into 76 (equation 39) was employed as the key step in total syntheses of the natural products ( )-A58365A and (C)-A58365B101.
O |
CO2 Et |
O |
CO2 Et |
|
N |
[O] |
N |
|
(37) |
||
|
|
||
|
|
|
|
|
|
MeOH |
|
|
|
|
OMe |
|
(71) |
|
(72) |
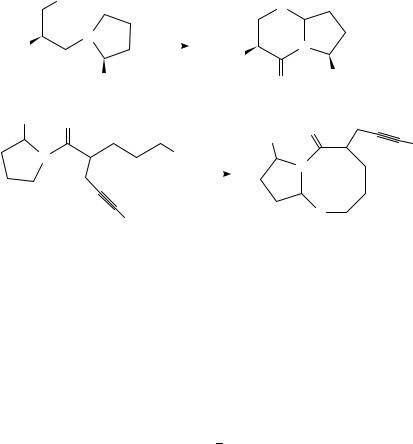
634 |
|
|
|
Albert J. Fry |
|
|
|
|||
|
OH |
|
|
|
|
O |
|
|
||
( |
)n |
|
|
|
( )n |
|
|
|||
|
|
|
|
|
|
|||||
|
|
|
N |
|
|
|
|
|
|
|
BocHN |
|
|
[O] |
|
N |
(38) |
||||
|
|
|
|
|
BocHN |
|
||||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
||
|
|
O |
|
|
|
|
|
CO2 Me |
|
|
|
|
|
CO2 Me |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
||
|
(73) |
|
|
|
|
|
(74) |
|
||
MeO2 C |
O |
|
|
|
|
|
O |
|
||
|
|
|
|
|
|
|
MeO2 C |
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
|
N |
OH |
|
|
||||||
|
|
N |
|
|||||||
( )n |
|
|
|
|
|
[O] |
|
|
(39) |
|
|
|
|
( |
)n |
||||||
|
|
|
|
|
||||||
|
|
|
Me |
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
(75) |
|
|
|
|
|
(76) |
|
||
C. Electroenzymatic Synthesis
There is much interest at the present time in the possibility of using electric current to regenerate the cofactors, especially NAD(P)C and NADH(P), required by enzymes which catalyze redox processes. If successful, this concept would combine the high degree of selectivity of the enzyme with the electrode as an inexpensive source of reducing (or oxidizing) power. The essential concept is illustrated in Scheme 20 for oxidation of an alcohol to the corresponding carbonyl compound. The electrode would oxidize the cofactor (in this case NADH) to the form (NADC ) needed for the enzymatic synthesis. After the desired conversion has been carried out, the NADH would return to the electrode to be oxidized again and begin the cycle anew. The idea is conceptually simple, but can be difficult to implement in practice. The very high cost of these redox cofactors (NADH costs more than $20,000/mole in bulk in 1996) requires that practical syntheses of this type exhibit turnover numbers of 1000 10,000 or higher. Furthermore, the reverse process, reduction of a carbonyl compound to an alcohol, generally fails because electrochemical reduction of NADC affords not NADH but an inactive dimer102. It is therefore necessary to employ an electrocatalyst to insure that NADC is converted to NADH at the electrode. This catalyst is typically a combination of a second enzyme, lipoamide dehydrogenase (LiDH; diaphorase), which catalyzes the NADC NADH interconversion, with an electron-transfer mediator, for example of the viologen (24) type103; certain organometallic complexes can effect the same conversion nonenzymatically104. Whitesides was one of the first to reduce these concepts to practice. His procedure105 for stereoselective L-lactate dehydrogenase (LDH)-catalyzed conversion of pyruvate to L-lactate involving passage of a current through a solution containing LiDH, LDH, 24, pyruvate and NADC (catalytic amount) is shown in Scheme 21. Other syntheses can be carried out by replacing pyruvate and LDH with the appropriate substrates and enzymes105.
Steckhan has reviewed progress in the area of electroenzymatic synthesis106. His review constitutes an excellent summary of previous work in this area. A few recent advances should be mentioned here. There is much interest in improving the practicality of these electroenzymatic conversions. Although Scheme 21 above proves the essential validity of the concept, it is highly impractical for large-scale operation because (a) the two enzymes
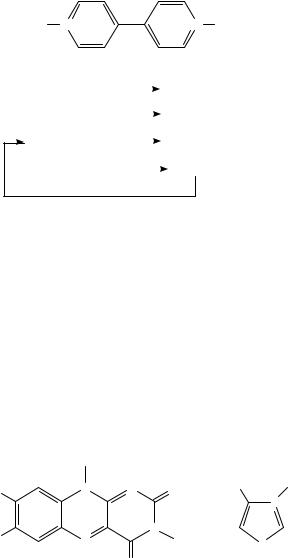
12. The electrochemistry of the CDC, CDO and CDN groups |
635 |
NADH 2e ! NADC C HC
NADC C R2CHOH ! NADH C R2CDO C HC
Net : R2CHOH 2e ! R2CDO C 2HC
SCHEME 20
H3 C |
+ |
|
|
|
|
+ |
CH3 |
2 X− |
|||
N |
|
|
|
|
|
|
N |
||||
|
|
(24) |
|
|
|
|
|
|
|||
|
|
242 + + e − |
|
|
|
241+ |
|
|
|
||
|
|
|
|
|
|
|
|
||||
2 241+ |
+ LiDH |
|
|
|
2 242 + + |
|
LiDH |
||||
|
|
|
|
||||||||
|
|
ox |
|
|
|
|
|
red |
|||
NAD+ |
+ |
LiDH |
|
|
|
|
LiDH |
|
+ |
NADH |
|
|
|
red |
|
|
ox |
|
|
||||
NADH |
+ |
pyruvate |
LDH |
|
|
NAD+ |
+ (+)-lactate |
||||
|
|
|
|||||||||
LiDH =lipoamide dehydrogenase; LDH =L-lactate dehydrogenase
SCHEME 21
are short-lived (half-lives less than two days), (b) viologen derivatives 24 are highly toxic and represent unacceptable potential contaminants, (c) the fact that all of the components are in solution makes separation of the desired product difficult and (d) any attempt to isolate the product will probably destroy the two redox enzymes. Any successful scheme for electroenzymatic synthesis must address all of these problems. One attractive approach is to replace the redox mediator LiDH with a synthetic catalyst. This is the approach taken by Steckhan, who uses a rhodium complex to convert NADC to NADH104,106. Similarly, Diederich has shown that the conversion of substituted benzaldehydes to methyl esters can be accomplished by electrochemical oxidation of a mixture of the aldehyde, a flavin derivative (77) and a thiazolium salt (78) in methanol (Scheme 22)107. The thiazolium salt first condenses with the aldehyde. The reduced form of the flavin (77red), a mimic of the redox enzyme active site, is oxidized at the anode to its active form (77ox), which then oxidizes the thiazolium adduct to an acyl thiazole, which in turn reacts with the solvent to afford the ester.
|
CH2 (CHOAc)3 CH2 OAc |
|
|
||
|
N |
N |
|
H C |
CH2 Ar |
H3 C |
O |
3 |
|
||
|
N |
||||
|
|
|
|
|
|
|
|
|
N |
|
Br− |
H3 C |
|
|
|
|
|
N |
|
CH3 |
|
S |
|
|
|
O |
|
|
|
|
(77) |
|
|
|
(78) |
SCHEME 22

636 |
Albert J. Fry |
An alternate approach to solving the problems (a) (c) cited in the previous paragraph seeks to retain the two enzymes and redox mediator 24 of Scheme 21, but somehow both extend the lifetime of the two enzymes and isolate them and also 24 from the bulk of solution so that separation of the product in a pure form, uncontaminated by 24, is easier. Substantial progress on this problem has recently been made by Fry and Sobolov and their collaborators. It was found that LiDH and 24 can be co-immobilized on a carbon electrode under an insoluble cation exchange membrane108,109, thus simplifying the composition of the electrolysis solution. A side benefit turned out to be a better than tenfold enhancement of the lifetime of LiDH when so immobilized. Unfortunately 24 slowly leaks into solution from the electrode under these conditions. This problem was addressed in two alternative ways: (a) chemically binding a viologen derivative to the enzyme110 and (b) incorporating LiDH and an oligomeric viologen derivative into a complex copolymer on the electrode surface111. The twin problems of the instability of LDH in solution and the difficulty of separating it from the electrolysis product were solved by resorting to the use of so-called ‘cross-linked enzyme crystals’112 or CLCs. The concept of a CLC is based on the facts that (a) enzymes retain their activity for long times in the crystalline state and that (b) components of a solution within which the crystal is placed can freely diffuse into and out of the crystal. An enzyme is crystallized from concentrated salt solution and the crystals are treated with a cross-linking agent, usually glutaraldehyde, which forms Schiff base linkages with lysine amino groups on the exterior of the crystal. The product is a CLC: an insoluble solid which retains the high activity of the original enzyme for very long times. Typically, CLCs have lifetimes measured in years in contrast with the lifetime (days) of the enzyme in solution. Fry and Sobolov and coworkers prepared CLCs of LDH113. These LDH-CLCs could be placed in a permeable pouch made of dialysis tubing. The enzyme is thus easily removed from the solution at the end of electrolysis for later re-use. All of these experimental variations could be incorporated into a single experiment in which an electrode coated with either the LDH-24 copolymer was immersed in a solution containing pyruvate, a trace of NADC , the LDH-CLC (in its dialysis pouch). Passage of current resulted in efficient production of L-lactate and ready isolation of the latter at the conclusion of electrolysis113. These concepts are readily incorporated into electrochemical flow cells designed for continuous electrolysis114.
Matsue’s group has immobilized both LiDH and alcohol dehydrogenase (ADH) on a single electrode115. The electrode was immersed in a solution containing methyl viologen (24), NADC and any of several cyclohexanone derivatives and current was passed. The ketones were reduced to the corresponding alcohols with good current efficiency. The course of the reduction is very similar to that of Scheme 21, except for the replacement of LDH and pyruvate by ADH and ketone and the fact that the viologen was in solution, not on the electrode. The efficiency of the process derives at least in part because the two enzymes are in close physical proximity; NADH and NADC merely have to shuttle back and forth between the two enzymes. A similar concept was introduced by Fry, Sobolov and coworkers, who co-crystallized the two enzymes LiDH and LDH and then crosslinked the resulting crystals to create cross-linked dual enzyme crystals116. These prove to be highly efficient for conversion of pyruvate to lactate, again because the two enzymes are in close physical proximity.
The characteristic stereospecificity of enzymes has been exploited in the design of an electrochemical cell for the conversion of L-lactate to D-lactate (Scheme 23)117. Enzymatic oxidation of L-lactate by L-lactate dehydrogenase affords pyruvate. Pyruvate is then reduced electrochemically to racemic lactate. A second enzymatic oxidation of the latter by L-lactate dehydrogenase selectively converts L-lactate to pyruvate, leaving D-lactate behind. The ingenious feature of this system is the fact that pyruvate can be re-reduced

12. The electrochemistry of the CDC, CDO and CDN groups |
637 |
|||||||
|
|
|
|
[O] |
|
|||
|
|
L-lactate |
|
|
|
|
pyruvate |
|
|
|
|
|
|
||||
|
|
|
|
L-lactate dehydrogenase |
|
|||
|
|
|
|
2 e −, 2H + |
|
|||
|
|
pyruvate |
|
L-lactate + D-lactate |
|
|||
|
|
|||||||
|
|
|
|
cathode |
|
|||
|
|
|
|
|
|
|
|
|
SCHEME 23
to racemic lactate, so that after several cycles one effects essentially complete overall conversion of one enantiomer to the other.
VI. ACKNOWLEDGMENTS
Financial support was provided by the National Science Foundation (Grant #CHE-94- 13128). Prof. Alan Bond and Mr. Richard Webster provided advance information concerning their studies on the electrochemistry of pyridine 2,6-dithioesters and related substances84.
VII. REFERENCES
1.A. J. Fry and R. G. Reed, in The Chemistry of Double-bonded Functional Groups: Supplement A. (Ed. S. Patai), Wiley, London 1977.
2.(a) H.-J. Schafer,¨ Top. Curr. Chem., 152, 91 (1990).
(b)B. C. I. Weedon, Adv. Org. Chem., 1, 1 (1960).
(c)A. K. Vijh and B. E. Conway, Chem. Rev., 67, 623 (1967).
3.(a) K. N. Campbell and E. E. Young, J. Am. Chem. Soc., 65, 965 (1943).
(b)L. Coche and J.-C. Moutet, J. Am. Chem. Soc., 109, 6887 (1987).
4.X. Cai and K. Kalcher, Electroanalysis, 6, 397 (1994).
5.(a) A. Bewick, D. E. Coe, G. B. Fuller and J. M. Mellor, Tetrahedron Lett., 21, 3827 (1980).
(b)A. Bewick, D. E. Coe, J. M. Mellor and D. J. Walton, J. Chem. Soc., Chem. Commun., 51 (1980).
(c)S. Torii, K. Uneyama and M. Ono, Tetrahedron Lett., 21, 2653 (1980).
(d)A. J. Bloom, M. Fleischmann and J. M. Mellor, Tetrahedron Lett., 25, 4971 (1984).
6.(a) R. H. Philp, Jr., R. L. Flurry and R. A. Day, Jr., J. Electrochem. Soc., 111, 328 (1964).
(b)M. E. Peover and J. D. Davis, J. Electroanal. Chem., 6, 46 (1963).
(c)G. H. Aylward, J. L. Garnett and J. H. Sharp, J. Chem. Soc., Chem. Commun., 137 (1966).
7.A. J. Fry, Synthetic Organic Electrochemistry, 2nd ed., Chap. 7, Wiley, New York, 1989.
8.H. Lund and M. M. Baizer, Organic Electrochemistry, 3rd ed., Chap. 6, Dekker, New York, 1991.
9.T. Shono, M. Ishifune, H. Kinugasa and S. Kashimura, J. Org. Chem., 57, 5561 (1992).
10.T. Shono, T. Nozoe, Y. Yamaguchi, M. Ishifune, M. Sakaguchi, H. Masuda and S. Kashimura,
Tetrahedron Lett., 32, 1051 (1991).
11.G. Farnia, F. Maruzzi, G. Melloni, G. Sandona and M. V. Zucca, J. Am. Chem. Soc., 111, 918 (1989).
12.(a) M. Fleischmann and D. Pletcher, Tetrahedron Lett., 6255 (1968).
(b)J. Bertram, M. Fleischmann and D. Pletcher, Tetrahedron Lett., 349 (1971).
13.J. Gora,´ K. Smigielski and J. Kula, Synthesis, 759 (1989).
14.A. J. Fry and G. S. Ginsburg, J. Am. Chem. Soc., 101, 3927 (1979).
¨
15. A. Dastan, U. Demir and M. Bala,´ J. Org. Chem., 59, 6534 (1994). 16. D. H. Geske, J. Am. Chem. Soc., 81, 4145 (1959).
17. T. Shono, T. Nozoe, H. Maekawa and S. Kashimura, Tetrahedron Lett., 29, 555 (1988). 18. T. Shono, H. Maekawa, T. Nozoe and S. Kashimura, Tetrahedron Lett., 31, 895 (1990). 19. E. Laurent, R. Tardivel and H. Thiebault, Tetrahedron Lett., 24, 903 (1983).
20. K. Suda and H. Ohmori, J. Chem. Soc., Chem. Commun., 1310 (1990). 21. M. Schmittel, J. Heinze and H. Trenkle, J. Org. Chem., 60, 2726 (1995).
638 |
Albert J. Fry |
22.M. Rock¨ and M. Schmittel, J. Chem. Soc., Chem. Commun., 1739 (1993).
23.M. Cariou and J. Simonet, Tetrahedron Lett., 32, 4913 (1991).
24.F. Barba, M. G. Quintanilla and G. Montero, J. Org. Chem., 60, 5658 (1995).
25.K. D. Moeller and L. V. Tinao, J. Am. Chem. Soc., 114, 1033 (1992).
26.L. V. Tinao-Wooldridge, K. D. Moeller and C. M. Hudson, J. Org. Chem., 59, 2381 (1994).
27.K. D. Moeller, M. R. Marzabadi, D. G. New, M. Y. Chiang and S. Keith, J. Am. Chem. Soc., 112, 6123 (1990).
28.H. Simon, H. Gunther,¨ J. Bader and W. Tischer, Angew. Chem., Int. Ed. Engl., 20, 861 (1981).
29.A. J. Fry, Synthetic Organic Electrochemistry, 2nd ed., Chap. 6, Wiley, New York, 1989.
30.A. J. Fry and R. G. Reed, J. Am. Chem. Soc., 91, 6448 (1969).
31.T. Shono, N. Kise, E. Shirakawa, H. Matsumoto and E. Okazaki, J. Org. Chem., 56, 3063 (1991).
32.T. Shono, N. Kise and E. Okazaki, Tetrahedron Lett., 33, 3347 (1992).
33.T. Shono, N. Kise, R. Nomura and A. Yamanami, Tetrahedron Lett., 34, 3577 (1993).
34.S. F. Martin, C.-P. Yang, W. L. Laswell and H. Rueger,¨ Tetrahedron Lett., 29, 6685 (1988).
35.A. Boulmedais, M. Jubault and A. Tallec, Tetrahedron, 45, 5497 (1989).
36.T. Shono, N. Kise, T. Fujimoto, A. Yamanami and R. Nomura, J. Org. Chem., 59, 1730 (1994).
37.A. Guirado, A. Zapata and M. Fenor, Tetrahedron Lett., 33, 4779 (1992).
38.T. Shono, Y. Matsumura, K. Tsubata, T. Kamada and K. Kishi, J. Org. Chem., 54, 2249 (1989).
39.A. J. Fry, Synthetic Organic Electrochemistry, 2nd ed., Wiley, New York, 1989, pp. 4 and 317.
40.A. Padwa, 1,3-Dipolar Cycloaddition Chemistry, 2 vols., Wiley, New York, 1984.
41.J.-i. Yoshida, M. Itoh, S.-i. Matsunaga and S. Isoe, J. Org. Chem., 57, 4877 (1992).
42.L. L. Limacher, F. D. Delay, N. Bedert´ and P. Tissot, Helv. Chim. Acta, 72, 1383 (1989).
43.T. Chiba and M. Okimoto, J. Org. Chem., 57, 1375 (1992).
44.M. Okimoto and T. Chiba, J. Org. Chem., 55, 1070 (1990).
45.H. Lund and M. M. Baizer, Organic Electrochemistry, 3rd ed., Chap. 3, Dekker, New York, 1991.
46.C. P. Andrieux, M. Grzeszchuk and J.-M. Saveant, J. Am. Chem. Soc., 113, 8811 (1991).
47.(a) K. Wiesner, Z. Elektrochem., 49, 164 (1943).
(b)K. Wiesner, Collect. Czech. Chem. Commun., 12, 64 (1947).
48.U. Nain and S. Kumbhat, J. Electrochem. Soc. India, 43, 63 (1994).
49.G. Pezzatini and H. Wei, Sci. Phys. Sci., 6, 1 (1994).
50.N. L. Weinberg, in Electroorganic Synthesis: Festschrift for Manuel M. Baizer (Eds. R. D. Little and N. L. Weinberg), Dekker, New York, 1991, p. 55.
51.Y. Ono, Y. Nishiki and T. Nonaka, Chem. Lett., 1623 (1994).
52.K. Matsuda, M. Atobe and T. Nonaka, Chem. Lett., 1619 (1994).
53.Y. Ikeda and E. Manda, Chem. Lett., 839 (1989).
54.E. Leonard, E. Dunach˜ and J. Perichon,´ J. Chem. Soc., Chem. Commun., 276 (1989).
55.F. Fournier, J. Berthelot and Y.-L. Pascal, Can. J. Chem., 61, 2121 (1983).
56.H. Schick, R. Ludwig, K.-H. Schwarz, K. Kleiner and A. Kunath, J. Org. Chem., 59, 3161 (1994).
57.Y. Rollin, S. Derien, E. Dunach,˜ C. Gebelhenne and J. Perichon,´ Tetrahedron, 49, 7723 (1993).
58.S. Durandetti, S. Sibille and J. Perichon,´ J. Org. Chem., 54, 2198 (1989).
59.M. Tokuda, N. Mimura, T. Karasawa, H. Fujita and H. Suginome, Tetrahedron Lett., 34, 7607 (1993).
60.(a) H. Hebri, E. Dunach˜ and J. Perichon,´ Tetrahedron Lett., 34, 1475 (1993).
(b)H. Hebri, E. Dunach˜ and J. Perichon,´ J. Chem. Soc., Chem. Commun., 499 (1993).
61.M. Makosza and K. Grela, Tetrahedron Lett., 36, 9225 (1995).
62.J. M. Tanko and R. E. Drumright, J. Am. Chem. Soc., 112, 5362 (1990).
63.M. Kimura, H. Yamagishi and Y. Sawaki, Denki Kagaku, 62, 1119 (1994).
64.T. Shono, S. Kashimura, Y. Mori, T. Hayashi, T. Soejima and Y. Yamaguchi, J. Org. Chem., 54, 6001 (1989).
65.E. Kariv-Miller, H. Maeda and F. Lombardo, J. Org. Chem., 54, 4022 (1989).
66.F. Lombardo, R. A. Newmark and E. Kariv-Miller, J. Org. Chem., 56, 2422 (1991).
67.S. Kashimura, M. Ishifune, Y. Murai, N. Moriyoshi and T. Shono, Tetrahedron Lett., 36, 5041 (1995).
68.A. S. C. Chan, T. T. Huang, J. H. Wagenknecht and R. E. Miller, J. Org. Chem., 60, 742 (1995).
69.N. Kise, T. Suzumoto and T. Shono, J. Org. Chem., 59, 1407 (1994).
12. The electrochemistry of the CDC, CDO and CDN groups |
639 |
70.(a) T. Shono and N. Kise, Tetrahedron Lett., 31, 1303 (1990).
(b)T. Shono, N. Kise, T. Fujimoto, N. Tominaga and H. Morita, J. Org. Chem., 57, 7175 (1992).
71.A. J. Fry, Synthetic Organic Electrochemistry, 2nd ed., Wiley, New York, 1989, p. 157.
72.H. Maeda, T. Maki and H. Ohmori, Denki Kagaku, 62, 1109 (1994).
73.G. T. Cheek and P. A. Horine, J. Electrochem. Soc., 131, 1796 (1984).
74.A. Guirado, F. Barba, C. Manzanera and M. D. Velasco, J. Org. Chem., 47, 142 (1982).
75.(a) G. A. Urove and D. G. Peters, Electrochim. Acta, 39, 1441 (1994).
(b)M. S. Mubarak, J. Electroanal. Chem., 394, 239 (1995).
76.A. Guirado, F. Barba and J. Martin, Synth. Commun., 13, 327 (1983).
77.J. H. P. Utley, Y. Gao and R. Lines, J. Chem. Soc., Chem. Commun., 1540 (1993).
78.(a) M. S. Mubarak, G. A. Urove and D. G. Peters, J. Electroanal. Chem., 350, 205 (1993).
(b)G. A. Urove and D. G. Peters, Tetrahedron Lett., 34, 1271 (1993).
(c)M. S. Mubarak and D. G. Peters, J. Electrochem. Soc., 142, 713 (1995).
79.J.-C. Folest, E. Pereira-Martins, M. Troupel and J. Perichon,´ Tetrahedron Lett., 34, 7571 (1993).
80.(a) M. Heintz, M. Devand, H. Hebri, E. Dunach˜ and M. Troupel, Tetrahedron, 49, 2249 (1993).
(b)S. Kashimura, Y. Murai, M. Ishifune, H. Masuda, H. Murase and T. Shono, Tetrahedron Lett., 36, 4805 (1995).
81.D. Pletcher and L. Slevin, J. Chem. Soc., Perkin Trans. 2, 2005 (1995).
82.T. Shono, H. Masuda, H. Murase, M. Shimomura and S. Kashimura, J. Org. Chem., 57, 1061 (1992).
83.D. Archier-Jay, N. Besbes, A. Laurent, E. Laurent, H. Stamm and R. Tardivel, Tetrahedron Lett., 30, 2271 (1989).
84.(a) R. D. Webster, A. M. Bond and T. Schmidt, J. Chem. Soc., Perkin Trans. 2, 1365 (1995).
(b)R. D. Webster, A. M. Bond and R. G. Compton, in press.
85.A. Guirado, A. Zapata and J. Galvez, Tetrahedron Lett., 35, 2365 (1994).
86.J. M. R. Mellado and M. M. Ruiz, J. Electroanal. Chem., 365, 71 (1994).
87.A. Guirado, F. Barba, M. Hursthouse and A. Arcas, J. Org. Chem., 54, 3205 (1989).
88.C. Zielinski and H. J. Schafer,¨ Tetrahedron Lett., 35, 5621 (1994).
89.A. J. Fry, Synthetic Organic Electrochemistry, 2nd ed., Chap. 9, Wiley, New York, 1989.
90.G. I. Nikishin, M. N. Elinson and I. V. Makhova, Angew. Chem., Int. Ed. Engl., 27, 1716 (1988).
91.M. Okimoto and T. Chiba, J. Org. Chem., 58, 6194 (1993).
92.T. Shono, Y. Matsumura, S. Katoh, T. Fujita and T. Kamada, Tetrahedron Lett., 30, 371 (1989).
93.S. Hara, S.-Q. Chen, T. Hatekeyama, T. Fukuhara, M. Sekiguchi and N. Yoneda, Tetrahedron Lett., 36, 6511 (1995).
94.T. Shono, T. Soejima, K. Takigawa, Y. Yamaguchi, H. Maekawa and S. Kashimura, Tetrahedron Lett., 35, 4161 (1994).
95.(a) J.-i. Yoshida, S.-i. Matsunaga and S. Isoe, Tetrahedron Lett., 30, 5293 (1989).
(b)J.-i. Yoshida, S.-i Matsunaga and S. Isoe, Tetrahedron Lett., 30, 219 (1989).
(c)J.-i. Yoshida, T. Murata and S. Isoe, Tetrahedron Lett., 27, 3373 (1986).
96.(a) J.-i. Yoshida, S.-i. Matsunaga and S. Isoe, Tetrahedron Lett., 30, 5293 (1989).
(b)J.-i. Yoshida, M. Itoh, S.-i. Matsunaga and S. Isoe, J. Org. Chem., 57, 4877 (1992).
97.M. Yamaguchi, Y. Tsukamoto and T. Minami, Chem. Lett., 1223 (1990).
98.(a) T. Shono, Electroorganic Chemistry as a New Tool in Organic Synthesis, Springer-Verlag, New York, 1984.
(b)A. J. Fry, Synthetic Organic Electrochemistry, 2nd ed., Chap. 8, Wiley, New York, 1989.
99.W. Li, C. E. Hanan, A. d’Avignon and K. D. Moeller, J. Org. Chem., 60, 8155 (1995).
100.U. Slomizynska, D. K. Chalmers, F. Cornille, M. L. Smythe, D. D. Beusen, K. D. Moeller and G. R. Marshall, J. Org. Chem., 61, 1198 (1996).
101.P. L. Wong and K. D. Moeller, J. Am. Chem. Soc., 115, 11434 (1993).
102.H. Jaegfeldt, Bioelectrochem. Bioenerg., 8, 355 (1981).
103.C.-H. Wong and G. M. Whitesides, Enzymes in Synthetic Organic Chemistry, Pergamon/Elsevier, Tarrytown, New York, 1994.
104.(a) R. Ruppert, S. Hermann and E. Steckhan, Tetrahedron Lett., 28, 6583 (1987).
(b)R. Ruppert, S. Hermann and E. Steckhan, Organometallics, 10, 1568 (1991).
105.R. DiCosimo, R. C. Wong, L. Daniels and G. Whitesides, J. Org. Chem., 46, 4622 (1981).
106.E. Steckhan, Top. Curr. Chem., 170, 83 (1994).
107.S.-w. Tam, L. Jimenez and F. Diederich, J. Am. Chem. Soc., 114, 1503 (1992).
640 |
Albert J. Fry |
108.A. J. Fry, S. B. Sobolov, M. D. Leonida and K. I. Voivodov, Tetrahedron Lett., 35, 5607 (1994).
109.A. J. Fry, S. B. Sobolov, M. D. Leonida and K. I. Voivodov, Denki Kagaku (Bull. Electrochem. Soc. Japan), 62, 1260 (1995).
110.K. I. Voivodov, S. B. Sobolov, M. D. Leonida and A. J. Fry, Bioorg. Med. Chem. Lett., 5, 681 (1995).
111.M. D. Leonida, A. J. Fry, S. B. Sobolov and K. I. Voivodov, Bioorg. Med. Chem. Lett., in press (1996).
112.N. L. St. Clair and M. A. Navia, J. Am. Chem. Soc., 114, 7314 (1992).
113.S. B. Sobolov, M. D. Leonida, A. Bartoszko-Malik, F. McKinney, J. Kim, K. I. Voivodov and A. J. Fry, J. Org. Chem., 61, 2125 (1996).
114.J. F. Fenton and A. J. Fry, unpublished research.
115. H.-C. Chang, T. Matsue and I. Uchida, in Electroorganic Synthesis: Festschrift for Manuel
M. Baizer (Eds. R. D. Little and N. L. Weinberg), Dekker, New York, 1991, p. 281.
116.S. B. Sobolov, A. Bartoszko-Malik, M. D. Leonida, K. I. Voivodov and A. J. Fry, in preparation.
117.A.-E. Biade, C. Bourdillon, J.-M. Laval, G. Mairesse and J. Moiroux, J. Am. Chem. Soc., 114, 893 (1992).