

Supplement F2: The Chemistry of Amino, Nitroso, Nitro and Related Groups.
Edited by Saul Patai Copyright 1996 John Wiley & Sons, Ltd.
ISBN: 0-471-95171-4
CHAPTER 10
Hydrogen bonding and complex formation involving compounds with amino, nitroso and
nitro groups
LUCIANO FORLANI |
|
|
Dipartimento di Chimica Organica ‘A. Mangini’, viale Risorgimento 4, |
|
|
40136-Bologna, Italy |
|
|
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
424 |
|
II. AMINO GROUP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
428 |
|
A. Hydrogen Bonding Interactions. Introduction . . . . . . . . . . . . . . . . . . |
428 |
|
B. Intermolecular Hydrogen Bonding . . . . . . . . . . . . . . . . . . . . . . . . . |
430 |
|
C. Self-association of Amines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
434 |
|
D. Intramolecular Hydrogen Bonding . . . . . . . . . . . . . . . . . . . . . . . . . |
435 |
|
E. Electron Donor-Acceptor Complexes . . . . . . . . . . . . . . . . . . . . . . . |
439 |
|
1. |
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
439 |
2. |
Intermolecular electron donor acceptor complexes . . . . . . . . . . . . |
440 |
3. |
Complexes between amines and halogens . . . . . . . . . . . . . . . . . . |
442 |
4. |
Intramolecular electron donor acceptor complexes . . . . . . . . . . . . |
443 |
III. NITROSO GROUP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
447 |
|
IV. NITRO GROUP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
450 |
|
A. Hydrogen Bonding . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
451 |
|
1. |
Intermolecular hydrogen bonding . . . . . . . . . . . . . . . . . . . . . . . |
451 |
2. |
Intramolecular hydrogen bonding . . . . . . . . . . . . . . . . . . . . . . . |
452 |
B. Electron Donor Acceptor Complexes . . . . . . . . . . . . . . . . . . . . . . |
455 |
|
V. COVALENT COMPLEXES BETWEEN AMINES AND NITRO |
|
|
ACTIVATED AROMATIC DERIVATIVES . . . . . . . . . . . . . . . . . . . . . |
457 |
|
423
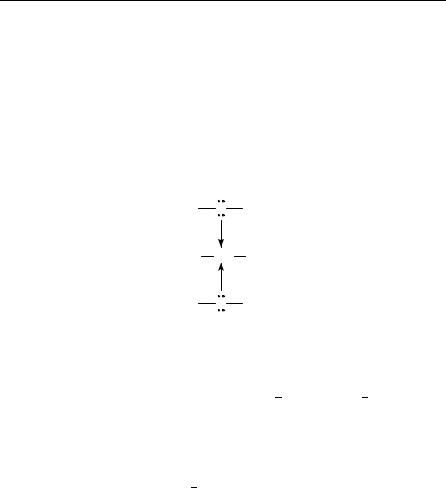
424 |
Luciano Forlani |
VI. NON-COVALENT INTERACTIONS BETWEEN AMINES
AND NITRO AROMATIC DERIVATIVES . . . . . . . . . . . . . . . . . . . . . 460
A.Nature of the Complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 461
B.Some Kinetic Features of Aromatic Nucleophilic
Substitution Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
465 |
VII. REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
470 |
I. INTRODUCTION
Although the ‘conventional’ way of describing reactions uses the chemical formula for ‘naked molecules’, this situation doesn’t really exist: every time a solute is dissolved in a solvent, solute/solvent and/or solute/solute interactions (of a greater or lesser degree) take place1. Consequently, the energy level of starting compounds interacting with solvents or solute (or self-associating) is different from that of the ‘free’ molecules.
In most text-books this point of view is scarcely considered, and few cases are reported. A frequent example is the stabilization of Grignard reagents by electron donor solvents (usually by lone pairs of the oxygen atoms of ethers), as shown in 1. This lack of attention is a simplification which generates little interest about the real nature of the starting materials and their energy levels, when reactions take place.
O
R Mg X
O
(1)
The first consequence is the problem of qualitatively or quantitatively rationalizing the effects of solvents and also of focusing on the physical and chemical properties of solvents, e.g. their polarity.
Thus, paying more attention to non-covalent solute solute or solute solvent interac-
tions in reacting systems (which may be investigated immediately after the mixing of the
˘ reagents) is a reasonable way to obtain useful analyses ˛ ˛K R ω of single and partic-
ular systems, as a starting point for a general view and rationalization (a synthesis, -˛K ω) of this problem.
Some empirical parameters describing interactions between solvents and solutes, or the dependence of the reactivities on changes of the solvent, are important for a general explanation of molecular interactions1 3 (in particular non-covalent interactions) because they introduce the idea that the immediate neighbours of the solute (or of the reaction centres) are a micro-region of great interest. Classical parameters which consider the medium as a homogeneous system may be hardly used to explain physical and chemical properties of solutes in solutions.
In 1948, Grunwald and Winstein2,4 attempted to define the ionizing power of a solvent by the Y parameter, based on the comparison of the rate for the solvolysis of t-butyl chloride. In 1956, Kosower5 made an attempt to define the polarity of a medium (solvent) by introducing the Z parameter based on the spectroscopic properties (in various solvents)

10. Hydrogen bonding and complex formation |
425 |
of the charge-transfer complex of 1-ethyl-4-carbomethoxypyridinium iodide, involving the transformation reported in Scheme 1.
COOCH3 |
COOCH3 |
|||||
|
|
J − |
hν |
|
|
J + e− |
|
|
|
|
|||
N + |
N |
|||||
C2 H5 |
C2 H5 |
|||||
SCHEME 1
In 1963, Dimroth6 introduced the ET parameter based on the spectroscopic properties of an intramolecular charge transfer of the pyridinium phenoxide betaine dyes 2, to define the solvent polarity for interpreting solvent effects on the reactivities.
Ph
Ph N + Ph
Ph |
Ph |
O −
(2)
In a recent review7 Reichardt listed the solvatochromic dyes used as indicators of the polarity of solvents.
Gutmann introduced3 the concepts of ‘donor number’ (donicity) and ‘acceptor number’ (acceptivity), as dimensionless numbers, for the characterization of donor properties of bases independently of the solvent.
The donor number has been defined as the negative H values for the formation of 1:1 adduct between solvents and antimony pentachloride, in 1,2-dichloroethane, at 25 °C8. In equilibrium 1, S is the solvent (with an unshared electron pair).
S |
C |
SbCl5 |
|
S |
|
SbCl5 |
1 |
|
|
|
|
|
|
In this way, the concept of donicity explains some properties of substances usually defined ‘apolar’ from their usual parameters of polarity (dielectric constant, dipolar moment, Et parameter value) but which presents high possibilities of interaction (and of solvatation) with positively charged centres. This is the case of tertiary amines such as triethylamine (or of ethers such as THF, dioxane) which shows usual polarity parameters near that of apolar solvents (benzene, chloroform, chlorobenzene, 1,2-dichloroethane, etc.) but high ability to coordinate positive charges.
426 |
Luciano Forlani |
The electrophilic properties of molecules were derived from 31P NMR chemical shifts9 produced by electrophilic solvents on triethylphosphine oxide as equilibrium 2 illustrates.
+ |
|
υ+ υ |
2 |
S Et3P O |
Et3 P O S |
||
C |
|
|
|
|
|
||
In this case the solvent (S) is an electron acceptor molecule.
In numerous systems, a fundamental role is played by the van der Waals forces10,11. Conformations of single molecules and intermolecular aggregations are strongly affected by van der Waals interactions which may be evaluated by quantum chemical methods. ‘True van der Waals’ interactions are those when the dominant attractive contribution is the dispersion energy12; the term ‘van der Waals complex’ may be considered better than ‘charge-transfer complex’ when no charge is transferred during the complex formation.
The solute/solvent interaction may be very important in determining the reactivity of solutes. An example arises from the tautomeric equilibria (in particular in heterocyclic series13, involving potential amino, hydroxy, mercapto groups; see Scheme 1) which may be shifted toward the left or right by simple changes of solvent or of polar solutes with consequent changes in spectroscopic properties and chemical behaviour14. Hydrogen bonds too are of fundamental importance in determining the position of the equilibrium of Scheme 2.
C XH |
C X |
N |
N |
H
X = O, S, NR
SCHEME 2
The synergism of hydrogen bond strength and -delocalization, as expressed by the idea of resonance assisted hydrogen bonding (RAHB)15, is an interesting tool in explaining some strong hydrogen bonds, and in particular the high availability to dimerization of linear16 or cyclic amides17, as well as 2-hydroxypyridine and all related substances including proteins and some bases of DNA. Tautomerism and geometrical properties of nitrogen containing heterocyclic bases bearing potential amino, hydroxy and mercapto groups are strictly related to the presence of dimers like 3.
N O
H H
O N
(3)
10. Hydrogen bonding and complex formation |
427 |
Non-covalent complexes (and hydrogen bonding complexes) are considered to be a relevant starting point in a conceptual revolution and in modern organic synthesis18; in general, hydrogen bonding is a fundamental force in a molecular recognition of biological macromolecules19,20.
Some practical applications of non-covalent interactions are also very interesting. The basis of the separation of enantiomers by the chromatographic method21 is the preferential interaction of one enantiomer of a substance with one enantiomer of another substance, which is usually part of the chiral stationary phase. Non-covalent interactions are more frequent: hydrogen bonding, host guest and donor acceptor interactions.
For example22, separation of enantiomers of chiral molecules (amines, alcohols, amino acids) is possible by using a chiral stationary phase obtained from 10-undecenyl esters of N-(2-naphthyl)-˛-amino acids (4). Amino groups are of fundamental importance in this practical application of the chiral recognition.
H |
O |
|
R |
N |
C O (CH2 )9 |
|
H |
(4)
Following essential early work on hydrogen bonding interactions23,24, the investigation of the interactions with protons have generated much interest and had a great deal of success. The proton (and, in general, the proton carriers) is a very particular acid species, because it is in an extreme position among the series of positive centres (for example, it is the least polarizable among the acids). Nevertheless, hydrogen bonding interactions are the most important (and the most investigated) non-covalent interactions, because of the diffusion of the problem of hydrogen bonding in both chemical and biochemical systems.
Comparing and making a distinction between hydrogen bonding and proton transfer can provide an important starting point in defining the field of hydrogen bonding interactions. In particular, in non-aqueous solvents, hydrogen bonding interactions may be related to aqueous acidities of both hydrogen donors and acceptors25. Attempts to rationalize the hydrogen-bond basicity (and hydrogen-bond acidity) using the equilibrium constant values of complexation, in tetrachloromethane, between a series of bases and some reference acids (or between a series of acids and a given base) afford scales of solute hydrogen-bond basicity (or acidity)26.
A simple (but essential) starting idea is that the conclusions arising from studies in different physical states (gas, liquid and solid) produce very important additional information, but it is dangerous to apply ‘sic et simpliciter’ the conclusions obtained in one state (in particular in solid) to the other states (in particular in solutions).
The relatively recent development27 of the direct methods of crystal structure analysis has produced a great increase in the number of crystal structures reported in the literature, particularly with regard to the possible hydrogen bonds (also for biological molecules). Hence, the classical spectroscopic data on hydrogen bonding in solution are backed up by X-ray diffraction analysis data.
As observed above, the solid state differs from the liquid state of solutions, under the usual experimental conditions, but some new ideas for an approach to hydrogen bonding originate from crystal analysis28. An example of this is offered by the fact that the hydrogen bonds observed in crystal structures are rarely linear. Bifurcated bonds (5,

428 |
Luciano Forlani |
6) and three-centre (7) (and four-centre) hydrogen bonds are frequently observed in the solid state (X is the hydrogen donor and Y is the hydrogen acceptor).
|
|
|
|
|
|
|
|
|
...Y |
|
|
|
|
|
|
|
|
|
. |
|
|
|
|
|
|
|
|
|
. |
|
|
|
|
|
|
|
|
H |
. |
H. |
|
|
|
|
|
....Y |
|
|
.... |
|
... |
|
||
|
|
. |
|
X |
|
.Y |
X |
|
..Y |
|
X H... |
|
|
|
|||||||
|
|
|
.... |
... |
|
|||||
. |
|
|
|
|
|
|
|
|||
|
|
|
. |
|
H |
|
H . |
|
|
|
|
|
|
Y |
|
|
|
|
|||
|
|
|
|
|
|
|
|
... |
|
|
|
|
|
|
|
|
|
|
|
. |
|
|
|
|
|
|
|
|
|
|
|
Y |
(5) |
|
|
(6) |
(7) |
|
|
||||
|
|
Bifurcated bonds |
|
Three-centre hydrogen bond |
||||||
The recognition of hydrogen bonds is an indispensable tool for designing molecular aggregates, in the field of crystal engineering29 as well as in the fields of supramolecular chemistry.
It is generally accepted that non-covalent interactions are the driving force of some biochemical transformations and interactions30,31. The molecular recognition, to form supramolecular species32, is possible if intermolecular interactions, non-covalent in character, originate in a well defined pattern33; consequently, the investigation of simple models of non-covalent interactions is the first step to understanding more complicated processes and situations. Defined structures involving self-assembled sub-units are formed by non-covalent interactions34 and, in particular, by hydrogen bonding interactions35.
II.AMINO GROUP
A.Hydrogen Bonding Interactions. Introduction
Attempts to organize the hydrogen bonding interactions are reported in numerous papers which provide classifications and tables for many substances, including amines. In principle, the energetic classification in two main groups36 provides a simple tool (together with some other parameters such as bond length, geometry etc.) with which to recognize various hydrogen bonding interactions between solutes and solvents, and between solutes and solutes:
(a) weak hydrogen bonding: bond energy <50 kJ mol 1 and mostly <30 kJ mol 1;
(b) strong hydrogen bonding: bond energy >50 kJ mol 1 and in some cases >100 kJ mol 1.
Hydrogen bonding versus proton transfer37 and the nature of the hydrogen bond38, both in the liquid and in the gas phase, are subjects of extensive investigations and useful empirical rules to draw hydrogen bond patterns are well codified for organic compounds39.
Attempts to evaluate the effective solute hydrogen bond acidity and basicity40 produce quantitative scales of solute hydrogen-bonding, which may be of interest for application also to biochemical processes41. The indicator pair 4-nitroaniline/4-nitro-N,N- dimethylaminoaniline are used to state the equivalence42 of solute and solvent scales of hydrogen bond basicity.
A scale of relative hydrogen bond basicity for a wide variety of solutes by means of their retention in gas chromatography was developed43 by using 4-dodecylphenyl-˛-˛- bis-(trifluoromethyl) methanol 8 as hydrogen bond donor. The comparison of results with those obtained by hydrogen bond basicity measures employing FT-IR methods indicates good agreement when the formation of hydrogen bond complexes is 1:1. In other cases discrepancies may be observed.
10. Hydrogen bonding and complex formation |
429 |
|
|
CF3 |
|
H25C12 |
C OH |
|
CF3
(8)
Usually, proton transfer in acid base equilibria is a fast process, but very hindered systems show slow kinetics. The equilibrium (equation 3) regarding 2,4-dialkylpyridines proceeds through locked-rotor and with intermediates of low entropy44.
|
+ |
|
+ N |
N |
N |
H |
|
|
(3)
+
N +
H
Many papers concerning the structural determination of hydrogen bonds in crystals were recently reviewed26. Hydrogen bonds are responsible for the self-assembling of
H
O N O
H H H H
N N
N N
H H
N N N N
O
H H H H
N N N N N N
H H H H H H
O N O O N O
H H
N
N N N N
H H H H
N N
O O
H H
N N N
H H
(9)
430 |
Luciano Forlani |
supramolecular aggregates45 and, together with other non-covalent interactions which control molecular recognition, also for other self-assembly phenomena46 and for the formation of hydrogen-bonded helices35 as well as for the non-covalent synthesis of aggregates47.
The cyclic hexamer 9 (named ‘rosette’) presents 18 hydrogen bonds48,49; 9 is the first step in the formation of the cyanuric acid/melamine lattice formed by a sheet of non-covalent aggregates50.
The hydrogen bond is the driving force in forming a supramolecular aggregate (with 54 hydrogen bonds51) between a compound containing nine melamine rings 10 and nine neohexylisocyanurate derivatives 11. These supramolecular aggregates are well defined compounds, as tested by 1H, 13C NMR spectroscopic data, by gel permeation chromatography and vapor pressure osmometry52. The size of the substituents on the melamine ring (and on the cyanuric ring) influences the preorganization of precursors and the stability of aggregates: the stability of rosettes decreases as the size of substituents is reduced53.
|
NH2 |
|
|
O |
|
N |
|
N |
N |
|
NH |
R1HN |
N |
NHR |
O |
N |
O |
|
|
|
|
H |
|
|
(10) |
|
|
(11) |
|
B. Intermolecular Hydrogen Bonding
In principle, for a given acid (AH) base (B) pair (such as nitrogen bases and the O H group), interactions between AH and B are controlled by the equilibrium (equation 5) between the molecular complex arising from a first equilibrium (equation 4) and the ion pair, including the solvation of the ion pair and the effect of the delocalization charge54.
A |
|
H |
C |
B |
|
A H |
B |
(4) |
|
|
|
|
|
|
Ð Ð Ð |
|
|||
A |
|
H |
|
B |
|
|
|
+ |
(5) |
|
Ð Ð Ð |
A |
Ð Ð Ð |
H B |
|||||
|
|
|
|
|
|
|
|||
Interactions between tertiary aliphatic amines or N,N-dialkylanilines and substituted phenols are generally reported as models of O HÐ Ð ÐN hydrogen bonds affording molecular complexes (equilibrium 4). A number of complexes between primary, secondary and tertiary aliphatic amines and dihydroxybenzenes (or dihydroxynaphthalenes) were isolated55 to investigate the stoichiometry of these complexes. The phenol/amine ratios observed included values of 1:1, 2:1, 3:1 and 3:2.
Complexes between amines and phenols in apolar solvents56 were extensively investigated by several techniques. The equilibrium between molecular complexes and ions was recently investigated57 by 1H NMR techniques for the complexes between phenols and N,N-dimethylaniline. The constant of the proton transfer equilibrium (K1 of equilibrium 7) increases on increasing pKa (D pKa of protonated base pKa of phenol) in water, and when the solutions are cooled.
ArOH C NR2Ar |
K |
ArO HÐ Ð Ð NR2Ar |
(6) |
|
|
||||
|
|
|
+ |
|
ArO HÐ Ð ÐNR2Ar |
K1 |
|
||
|
ArO Ð Ð ÐH NR2Ar |
(7) |
||
|
|
|
|
|

10. Hydrogen bonding and complex formation |
431 |
Triethylamine is a popular substance (because it is an aprotic base) to investigate interactions with OH groups, both aliphatic and aromatic, including diols: when the two OH groups are separated by a long chain they complex triethylamine independently58. Mixtures of triethylamine and phenols (investigated by vapour pressure osmometry) show non-ideal behaviour arising from the formation of complexes involving the presence of multimers12. Usually the formation constants of hydrogen-bridged complexes between phenols and bases are related to the protonation constant59 of bases60 but some deviations are observed (for example in the case of heterocyclic bases61), and they are explained by the particular nature of the considered bases.
The stability constant of complexes between ˇ-cyclodextrine and p-nitroaniline is higher than that of aniline because the resonace charge delocalization (and London dispersion interactions) is an important factor influencing the stability of these complexes62. This behaviour parallels that of corresponding phenols.
The dipole moments were related to hydrogen bond strength and to the ability of the proton acceptors (compounds with phosphoryl63 and carbonyl64 groups) to associate with phenol. Hydrogen bonded complexes of pyridines, quinolines and acridine with phenol are studied, in carbon tetrachloride, by dipole moment measures65. Dipole moments of the complexes were related to the ability of the acceptors to associate with phenol and with their basicity. Hydrogen bonds may either reinforce the dipole moments of the resultant of the group moments or may oppose the dipole moment direction, depending on the nature of the acceptor.
Amino groups may act not only as proton acceptor, but also as proton donor. Acidic N H protons interact with basic solvents. In these cases an ortho-nitro group in an aniline system competes with the solvent by an internal hydrogen bond66, as depicted in 12. The stretching frequencies (by IR spectra in carbon tetrachloride) of NH of complexes between N-methylaniline or diphenylamine (and some nitro-anilines66) and solvents depend on the proton accepting ability of the solvent (which is a moderate base)67. The frequency shifts are linearly related to the solvent’s donor number (DN)3.
The biological interest of hydrogen bonding in aqueous solutions is well known; protonated amines and phenolate ions in water are associated by hydrogen bonds. The association constant (K D 0.81 mol 1 dm3, measured in buffered aqueous solution) for the complex of the dication of ethylenediamine with the phenolate ion agrees with a weak hydrogen bonding N HÐ Ð ÐO68.
R |
H |
|
|
|
N |
O − |
|
|
|
|
N + |
Me |
|
H |
|
|
O |
|
|
|
|
r |
|
|
|
|
Me N |
H O |
|
|
|
|
||
|
|
Me |
|
|
|
(12) |
|
(13) |
|
The trimethylamine/water system (for 10 isotopomers) was investigated by microwave spectroscopy69. The analysis of the rotational constants indicate a structure with an essentially linear single hydrogen bond (r D 1.818 A˚ in 13).
The 13C resonances in derivatives of aniline reveal that the carbon bearing a partially deuterated NH2 group appears as a multiplet because of the deuterium isotope effect on the 13C chemical shift, when the hydrogen exchange is low. This effect is larger for

432 |
Luciano Forlani |
systems with intramolecular hydrogen bonds and allows an evaluation of the energies of the hydrogen bonds70. These observations are extended to intermolecular hydrogen bonds in partially deuterated nucleoside systems and indicate base-pairing of the nucleosides through intermolecular hydrogen bonds.
The H/D isotopic exchange in the uridine system (from starred nitrogen) may be explained by the mechanism (equation 8) involving dimerization of the hydroxy heterocycle70. Dimerization of hydroxy derivatives of aza-containing heterocycles plays an important role in assembling molecules, and in their tautomeric equilibria.
|
|
|
O |
R |
|
|
|
O |
R |
|
|
|
|
N |
|
|
|
|
N |
|
O |
D |
N* |
|
|
O |
D |
N* |
|
|
N |
H |
O |
|
|
N |
H |
O |
|
|
N |
|
|
|
N |
|
|
|
|
R |
O |
|
|
|
R |
O |
|
|
(8) |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
O |
R |
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
N |
D + |
H |
N* |
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
R |
O |
|
|
O |
|
|
|
|
|
|
|
|
|
|
The dynamic 1H NMR technique allows investigations on the rate of exchange between 3-substituted quinuclidinium ions and water. The rate of dissociation of amine/water (or amine/alcohol) complexes is determined71 by the free energy contribution from the pKa- dependent hydrogen bond breaking, and from dispersion forces between acceptor and donor which may be at the most 40% of the activation energy of the dissociation of the complex. Similar importance may be attributed to a term for the formation of a cavity prior to the dissociation of the complexes.
Some para-substituted anilines in strongly hydrogen bonding solvents show two nonequivalently hydrogen bonded species with differently hybridized aniline nitrogens72; rehybridization of aromatic amino nitrogens depends on the OH acidity of the solvent molecule and the basicity of the substituted anilines. 14 shows three possible modes of interaction of p-aminoacetophenone and acohol, where the amino group is simultaneously both a proton donor and an acceptor.
R |
CH3 |
CH3 |
||||
|
N |
C |
|
|
|
|
|
|
. |
|
|
|
|
|
..... |
. |
|
|
|
|
O |
. |
O |
|
|
|
|
|
H . |
|
|
|
||
|
|
. |
.. |
|
|
|
|
|
H |
|
.. |
|
|
|
|
|
. |
|
|
|
H |
|
|
OR |
H |
|
OR |
|
|
|
||||
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
(14) |
|
|
|