

Гончуков Флюоресцентная диагностика
.pdfОбоснованно перспективным считается метод флюоресцентной спектроскопии, поскольку является чувствительным и недорогим. Он находит широкое применение в медицине при диагностике самых различных заболеваний. Хотя флюоресценция по интенсивности меньше возбуждающего излучения на три-четыре порядка, она легко детектируется и очень информативна. При этом для возбуждения флюоресценции можно использовать как лазеры, так и нелазерные источники света низкой интенсивности. Поэтому такой метод является еще и неинвазивным.
Флюоресцентная диагностика в стоматологии базируется на анализе спектров флюоресценции тканей зуба в норме и при патологиях. Основное вещество, из которого состоят ткани зуба, – гидроксиапатит. Гидроксиапатит и другие кристаллы входят в состав зубного камня. Это диэлектрики, которые, как известно, в чистом виде не флюоресцируют. Однако наличие примесей и дефектов приводит к их флюоресценции. Микроорганизмы, органические вещества, продукты распада также дают характерные спектры флюоресценции. Структура и химический состав твердой ткани зависят от многих факторов, включая возраст, пол, расу, социальное положение, традиционное питание, курение, хронические заболевания и т.д. В результате спектры флюоресценции могут существенно отличаться у пациентов. Детальный анализ спектров позволяет выбрать оптимальные для диагностики длины волн возбуждения и регистрации флюоресценции.
В фотобиологии различают эндо- и экзогенную флюоресценцию. Эндогенная флюоресценция или аутофлюоресценция присуща нативному состоянию исследуемого объекта, а экзогенная – объекту после его окрашивания красителями. Как показывают последние исследования, для достоверной флюоресцентной диагностики стоматологических заболеваний не требуется применение дополнительных красителей. Поэтому, для краткости, термин «аутофлюоресценция» мы заменяем флюоресценцией. Эндогенные флюорофоры весьма многочисленны. В табл. 3.1 приведены характерные длины волн возбуждения и излучения флюоресценции основных биологических молекул.
11
|
|
|
Таблица 3.1 |
|
|
|
|
|
|
|
Длина волны |
Длина волны |
|
|
Молекула |
возбуждения, |
флюоресцен- |
Происхождение |
|
|
нм |
ции, нм |
|
|
Триптофан |
275 |
350 |
Белки |
|
|
|
|
|
|
Коллаген |
335 |
390 |
Соединительная |
|
биоткань |
|
|||
|
|
|
|
|
|
|
|
|
|
Эластин |
360 |
410 |
Соединительная |
|
биоткань |
|
|||
|
|
|
|
|
|
|
|
|
|
Порфирины |
405, 540, 630 |
635 |
Микроорганизмы |
|
|
|
|
|
|
Возможности возбуждения и регистрации флюоресценции зависят от степени прохождения излучения через твердые ткани в рассматриваемом оптическом диапазоне длин волн. Процессы поглощения и рассеяния света в эмали и дентине существенно отличаются. Эмаль является более прозрачной тканью, чем дентин. На длине волны 633 нм коэффициенты поглощения и рассеяния эмали равны 0,97 и 1,1 см−1, в то время как у дентина эти коэффициенты равны 6,0 и 1200 см−1 соответственно. Основными рассеивателями света являются кристаллы гидроксиапатита, а поглотителями – органические молекулы. При переходе в ультрафиолетовый диапазон коэффициенты поглощения и рассеяния возрастают настолько, что глубина проникновения излучения в твердую ткань не превышает 0,1-0,3 мм. Для примера на рис. 3.1 приведен спектр поглощения дентина в диапазоне от 300 до 3000 нм. Таким образом, вклад во флюоресценцию от основных флюорофоров, кроме порфиринов, будет отражать изменения на поверхности биотканей. Порфирины же эффективно возбуждаются и излучают в видимом диапазоне длин волн. Они должны хорошо детектироваться в спектрах флюоресценции в присутствии микроорганизмов, поскольку содержатся преимущественно в митохондриях их клеток и продуктах жизнедеятельности.
12
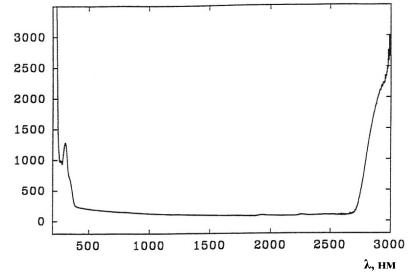
Рис. 3.1. Спектр поглощения дентина (см–1) в диапазоне от 300 до 3000 нм
4. ЯВЛЕНИЕ ФЛЮОРЕСЦЕНЦИИ
Согласно квантовой механике малые частицы (атомы, молекулы, ионы) могут принимать и отдавать энергию дискретными порциями – квантами. Изменению энергии соответствуют переходы между энергетическими состояниями (уровнями), структура которых индивидуальна для данной элементарной системы частицы. В зависимости от характера взаимодействия в системе энергетический спектр может быть дискретным (как у осциллятора), непрерывным (как у свободной частицы) либо смешанным (как, например, у возбужденных атомов).
Полная потенциальная энергия молекулы E (без учета ее движения как целое) складывается из энергии электронного возбуждения Eэ, энергии колебания Eк и энергии вращения Eв молекулы:
E = Eэ + Eк + Eв. |
(1) |
Для величин этих энергий выполняется примерное соотношение:
13
Eэ : Eк : Eв = 1 : (m/M)1/2 : m/M = |
(2) |
= 1-10 эВ : 10–2-10–1 эВ : 10–5-10–3 эВ, |
где m и M – массы электрона и молекулы соответственно.
Вотличие от свободных атомов потенциальная энергия молекулы зависит от расстояния r между ядрами молекулы. Для многоатомной молекулы энергетический спектр представляет собой поверхность в многомерном пространстве. Для двухатомной молекулы он имеет вид, представленный на рис. 4.1,а. Здесь приведены только основное и два нижних возбужденных электронных состояния. Каждому электронному состоянию, вид которого определяется внутримолекулярным взаимодействием, соответствует своя система колебательных уровней, а каждому колебательному – своя система вращательных уровней. Последние на рисунке не показаны, так как их энергии малы.
Минимум потенциальной энергии соответствует равновесному, наиболее вероятному расстоянию между ядрами, определяемому равенством между силами притяжения (dE/dr > 0) и отталкивания (dE/dr < 0). Для основного электронного состояния это расстояние
обозначено на рис. 4.1,а как r0. С ростом электронного возбуждения равновесное расстояние немного увеличивается, так как внут-
римолекулярная связь ослабляется. При увеличении Eк горизонтальные отрезки в пределах электронного состояния подымаются вверх и их размер растет, поскольку растет «размах» колебаний молекулы.
Вотсутствие возбуждения молекула старается занять самое низкое по энергии состояние. В реальных условиях положительной температуры (T > 0) вероятность перехода молекул в возбужденное состояние за счет тепловой энергии определяется распределением Больцмана:
N = N0 exp (– E/kT), |
(3) |
где N и N0 – число молекул в возбужденном и основном состояниях; E – разность энергий этих состояний; k – постоянная Больцмана. Если по той или иной причине молекулы перешли в неравновесное состояние, то тепловое равновесие с окружающей средой
14
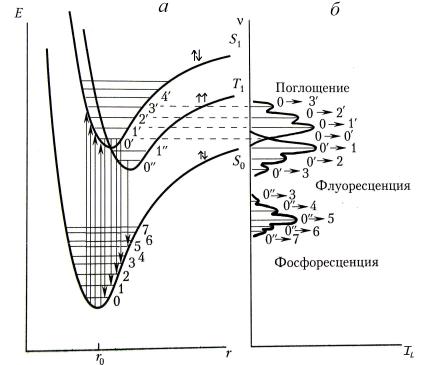
устанавливается в течение следующих 10–12 с и распределение молекул по энергетическим уровням определяется формулой (3).
Рис. 4.1. Схема формирования энергетических состояний (а) и спектров поглощения, флюоресценции и фосфоресценции (б)
Согласно принципу Франка–Кондона за время электронного перехода (~ 10–15 с) не происходит заметного изменения в положении ядер. Поэтому переходы между электронными состояниями можно изображать вертикальными стрелками, которые, очевидно, должны начинаться и заканчиваться в местах максимальной вероятности нахождения ядер. Из-за колебательной релаксации молекула очень быстро занимает низший в пределах данного электронного состояния уровень. При возбуждении молекулы ее колебания в пределах соответствующего горизонтального отрезка можно описать синусоидой. Поэтому для возбужденных колебательных уровней наи-
15
большая вероятность нахождения ядер соответствует местам касаний горизонтальных отрезков с кривой электронного состояния, а для основного колебательного уровня – вблизи их равновесного положения. Электронные переходы как снизу вверх (поглощение), так и сверху вниз (испускание) представлены на рис. 4.1,а соответствующими вертикальными стрелками.
Перевод молекулы в возбужденное, т.е. неравновесное, состояние возможен разными способами (рентгеновскими лучами, быстрыми частицами, электрическим полем, с помощью химической реакции и др.). В нашем случае будет рассматриваться возбуждение светом – фотовозбуждение. Диссипация полученной избыточной энергии может быть как безызлучательной (перенос энергии на молекулярное окружение, колебательная релаксация, внутренняя конверсия между электронными состояниями, переход в кинетическую энергию), так и излучательной. Во втором случае происходит преобразование поглощенной энергии в собственное излучение. Общее название этого явления – люминесценция – излучение, представляющее собой избыток над тепловым излучением тела и продолжающееся в течение времени, значительно превышающего период световых колебаний. Понятие явления люминесценции применимо к атомам и молекулам, находящимся в состоянии, близком к равновесному. Заметим, что в видимом диапазоне спектра тепловое излучение становится заметным при температурах ≥103 К, а люминесценция может наблюдаться при любых температурах. Поэтому люминесценцию называют еще холодным свечением.
По механизмам элементарных процессов различают резонансную, спонтанную и вынужденную люминесценцию. При резонансной люминесценции поглощение и испускание происходит между одними и теми же уровнями, и длины волн люминесценции и возбуждения совпадают λL = λexc. Наиболее часто наблюдается спонтанная люминесценция, когда часть энергии растрачивается безызлучательно на тепло. Тогда уровень испускания лежит ниже уровня возбуждения и λL > λexc. Это стоксова люминесценция. Возможен также менее вероятный процесс, когда молекула получает дополнительную энергию от теплового движения или от других молекул. Тогда λL < λexc, и люминесценция называется антистоксовой. Вынужденная люминесценция является более сложным процессом, в
16
который включен переход на промежуточный метастабильный уровень и требуется дополнительная энергия для перехода на уровень испускания. Такой вид люминесценции наблюдается в сложных молекулах при низкой температуре.
Традиционно принято разделять люминесценцию на флюоресценцию и фосфоресценцию. Под флюоресценцией понимается излучение, быстро (за ~10–9 с) затухающее после прекращения возбуждения. Фосфоресценцией считается свечение, продолжающееся от микросекунд до секунд и более после возбуждения. Такое разделение устарело и носит чисто качественный характер. Длительность люминесценции определяется временем жизни возбужденного состояния.
Из закона сохранения энергии следуют правила отбора, которые накладывают запрет на некоторые переходы. Для электронных переходов правила отбора связаны с мультиплетностью 2S + 1 (S – спиновое квантовое число) и запрещают переходы с изменением спина. Мультиплетность характеризует число возможных ориентаций в пространстве полного спина атомной системы. При 2S + 1 = = 1, 2, 3, 4, … уровни энергии называют синглетными, дублетными, триплетными, квартетными и т. д. Таким образом, приведенные на рис. 4.1,а синглет-синглетные переходы разрешены, а триплетсинглетные переходы запрещены. Основное состояние почти всех молекул с четным числом электронов таково, что молекулярные орбитали заполнены парами электронов. Согласно принципу Паули такие пары электронов должны иметь противоположно направленные спины. Следовательно, основное состояние является синглетным, так как S = 0. Реально запрет на переходы с изменением спина снимается благодаря спин-орбитальному взаимодействию. Однако из-за запрета вероятность такого перехода мала, триплетный уровень на рис. 4.1,а является метастабильным и его распад происходит медленно. Излучение при триплет-синглетных переходах и есть фосфоресценция (рис. 4.1,б).
В данном издании рассматриваем возможность использования оптического метода диагностики, когда воздействующее и регистрируемое излучения относятся к видимой и ближней ультрафиолетовой области спектра. Связь изменения энергии квантовой системы с излучением при поглощении и испускании определяется формулой Планка
17
E = hν, |
(4) |
где h – постоянная Планка; ν – частота электромагнитных колебаний. Учитывая энергию кванта излучения (1-3 эВ) и соотношение (2), процессы поглощения и излучения должны соответствовать переходам между электронными состояниями, как показано на рис.4.1,а. Энергетические уровни молекулы имеют свою индивидуальную структуру и спектроскопические параметры, что определяет характерный вид ее спектров поглощения и испускания.
Для каждого перехода спектральная линия поглощения или излучения имеет конечную ширину. Механизмы уширения спектральных линий разнообразны. Естественное или радиационное уширение определяется конечным временем жизни элементарной частицы в возбужденном состоянии. Меньше радиационного уширение быть не может. Процессы теплового движения частицы и ее взаимодействия с окружающими частицами и кристаллической решеткой дают дополнительный, обычно основной, вклад в уширение. Для конденсированных сред, в том числе и для биологических тканей, ширина спектральной линии составляет, как правило, около 10% от центральной частоты. Поэтому у молекулы спектральные линии, связанные с переходами между близко расположенными колебательными уровнями электронных состояний, полностью или частично, как показано на рис. 4.1,б, перекрыты.
В формировании спектров флюоресценции имеются свои особенности. После поглощения кванта света и быстрой колебательной релаксации испускание флюоресценции всегда происходит с нижнего колебательного уровня возбужденного состояния. Таким образом, спектр флюоресценции не зависит от длины волны возбуждающего излучения (правило Каши). Другая особенность связана с тем, что структура и вероятности переходов на колебательные уровни близки у молекулы в основном и возбужденном электронном состоянии. Иными словами, структурная организация молекулы в основном и возбужденном состояниях мало отличается. Следствием этого является зеркальная симметричность спектров поглощения и стоксовой флюоресценции относительно длинноволновой границы поглощения (правило Левшина). Эта особенность хорошо проявляется на рис. 4.1,б при сравнении спектров поглощения и флюоресценции на шкале частот.
18
Спектр флюоресценции несет важную информацию о структуре и химическом составе исследуемого объекта. Выбор оптимальных длин волн возбуждения и регистрации флюоресценции позволяет идентифицировать патологические изменения в тканях зуба на ранних стадиях развития заболеваний.
5. ПРАКТИЧЕСКОЕ ПРИМЕНЕНИЕ ФЛЮОРЕСЦЕНЦИИ
5.1. Источники возбуждения
Для фотовозбуждения флюоресценции можно применять различные источники света. Чтобы свет эффективно поглощался молекулой, длина волны излучения должна совпадать с той или иной спектральной линией поглощения. К ширине спектра возбуждающего излучения жестких требований не предъявляется, но она не должна пересекать длинноволновую границу спектра поглощения молекулы. В противном случае трудно детектировать слабую стоксову флюоресценцию на фоне интенсивного возбуждающего излучения. Поэтому на практике используемые мощные лампы снабжаются светофильтрами или монохроматорами.
В последнее время для возбуждения флюоресценции широко используются лазеры и светодиоды. По своим пространственным и яркостным характеристикам, а также монохроматичности (5-15 нм) светодиоды, конечно, уступают лазерам. Однако технология изготовления светодиодов непрерывно совершенствуется. Они имеют малые габариты, низкую цену и высокую долговечность. Весьма существенно, что в отличие от лазеров у светодиодов имеется возможность широкого выбора длин волн излучения.
Тем не менее лазеры не уступают своих позиций. Узкий спектр излучения (~ 10–2 нм) лазеров позволяет довольно просто отсечь их излучение от флюоресценции. Кроме того, поскольку лазерное излучение узконаправленное, а спонтанное излучение флюоресценции «светит» в 4π стерадиан, то регистрация флюоресценции под углом дает дополнительную возможность для ослабления засветки от возбуждающего излучения. Что касается цены и габаритов, то многие приемлемые для работы маломощные лазеры могут быть вполне сравнимы со светодиодами.
19
Далее приводятся результаты наших исследований, в которых для возбуждения флюоресценции применялись газовые, твердотельные и полупроводниковые непрерывные лазеры, а также светодиоды. Излучение источников в совокупности охватывало диапазон от 337 до 658 нм. Мощность их выходного излучения не превышала 10 мВт.
5.2.Волоконно-оптический спектрометр ЛЭСА
Кнастоящему времени разработан ряд удобных спектрометров, адаптированных к применению в биофотонике. Мы используем здесь результаты наших исследований, выполненных с помощью одного из них – волоконно-оптического спектрометра ЛЭСА-5 (ЗАО «Биоспек», Россия). Действие спектрометра основано на пространственном разложении исследуемого излучения по длинам волн с помощью дифракционной решетки и регистрации этого излучения на ПЗС линейке. Работа спектрометра поддерживается специальным программным обеспечением. Спектры регистрируются в реальном времени и отображаются на экране монитора компьютера. Спектральное разрешение около 8 нм, рабочий диапазон от
290 до 1100 нм.
Как во всяком спектральном приборе, регистрируемый спектр I'(λ) отличается от исходного спектра I(λ), поскольку коэффициент передачи K(λ) прибора зависит от длины волны:
I'(λ) = I(λ)ּK(λ). |
(5) |
Зависимость коэффициента передачи от длины волны обусловлена соответствующими зависимостями у всех оптических и фотоэлектронных составляющих спектрометра. В первую очередь это касается ПЗС линейки и дифракционной решетки. Весьма существенным здесь является интерференция анализируемого излучения на тонком просветляющем покрытии ПЗС линейки, которая приводит к значительной периодической модуляции регистрируемого спектра по амплитуде.
Неравномерность коэффициента передачи спектрометра можно учесть с помощью вспомогательного опорного источника с известным спектром излучения I0(λ). На практике в качестве такого ис-
20