

Berrar D. et al. - Practical Approach to Microarray Data Analysis
.pdf164 |
Chapter 8 |
the analysis of the learned Bayesian network could generate some interesting hypotheses which could guide further biological experiments.
ACKNOWLEDGMENTS
This work was supported by the BK-21 Program from Korean Ministry of Education and Human Resources Development, by the IMT-2000 Program and the NRL Program from Korean Ministry of Science and Technology.
REFERENCES
Alon U., Barkai N., Notterman D.A., Gish K., Ybarra S., Mack D., Levine A.J. (1999). Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci USA 96(12):6745-50
Bang J.-W., Gillies D. Using Bayesian networks with hidden nodes to recognize neural cell morphology (2002). Proc of the Seventh Pacific Rim International Conference on Artificial Intelligence; 2002 August 18 - 22; Tokyo. Heidelberg: Springer-Verlag (to appear).
Ben-Dor A., Bruhn L., Friedman N., Nachman I., Schummer M., Yakhini Z. (2000). Tissue classification with gene expression profiles. J Comput Biol 7(3/4):559-84
Chickering D.M. (1996). “Learning Bayesian Networks is NP-Complete.” In Learning from Data: Artificial Intelligence and Statistics V, Doug Fisher, Hans-J. Lenz, eds. New York, NY: Springer-Verlag.
Cooper G.F. (1990). The computational complexity of probabilistic inference using Bayesian belief networks. Artificial Intelligence 42(2-3):393-405
Cover T.M., Thomas J.A. (1991). Elements of Information Theory. NY: John Wiley & Sons.
Dempster A.P., Laird N.M., Rubin D.B. (1977). Maximum likelihood from incomplete data via the EM algorithm (with discussion). J R Stat Soc Ser B 39(1): 1-38
Dougherty J., Kohavi R., Sahami M. (1995). Supervised and unsupervised discretization of continuous features. Proc of the Twelfth International Conference on Machine Learning; 1995 July 9 - 12; Tahoe City. San Francisco: Morgan Kaufmann Publishers.
Dubitzky W., Granzow M., Berrar D. (2002). “Comparing Symbolic and Subsymbolic Machine Learning Approaches to Classification of Cancer and Gene Identification.” In Methods of Microarray Data Analysis, Simon M. Lin, Kimberly F. Johnson, eds. Norwell, MA: Kluwer Academic Publishers.
Friedman N., Geiger D., Goldszmidt M. (1997). Bayesian network classifiers. Machine Learning 29(2/3):131-63.
Friedman N., Goldszmidt M. (1999.) “Learning Bayesian Networks with Local Structure.” In Learning in Graphical Models, Michael I. Jordan, ed. Cambridge, MA: MIT Press, 1999.
Friedman N., Linial M., Nachman I., Pe’er D (2000). Using Bayesian networks to analyze expression data. J Comput Biol 7(3/4):601-20.
Golub T.R., Slonim D.K., Tamayo P., Huard C., Gaasenbeek M., Mesirov J.P., Coller H., Loh M.L., Downing J.R., Caligiuri M.A., Bloomfield C.D., Lander E.S. (1999). Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286(5439):531-7.
8. Bayesian Network Classifiers for Gene Expression Analysis |
165 |
Hartemink A.J., Gifford D.K., Jaakkola T.S., Young R.A. (2001). Combining location and expression data for principled discovery of genetic regulatory network models. Proc. Seventh Pacific Symposium on Biocomputing; 2002 January 3 - 7; Lihue. Singapore: World Scientific Publishing.
Haykin S. (1999). Neural Networks – A Comprehensive Foundation, 2nd edition. NJ: PrenticeHall.
Heckerman D., Geiger D., Chickering D.M. (1995). Learning Bayesian networks: the combination of knowledge and statistical data. Machine Learning 20(3):197-243
Heckerman D. (1999). “A Tutorial on Learning with Bayesian Networks.” In Learning in Graphical Models, Michael I. Jordan, ed. Cambridge, MA: MIT Press.
Herblot S., Apian P.D., Hoang T. (2002). Gradient of E2A activity in B-cell development. Mol Cell Biol 22:886-900
Hwang K.-B., Cho D.-Y., Park S.-W., Kim S.-D., Zhang B.-T. (2002). “Applying Machine Learning Techniques to Analysis of Gene Expression Data: Cancer Diagnosis.” In Methods of Microarray Data Analysis, Simon M. Lin, Kimberly F. Johnson, eds. Norwell, MA: Kluwer Academic Publishers.
Jensen F.V. Bayesian Networks and Decision Graphs. NY: Springer-Verlag, 2001.
Khalidi H.S., O’Donnell M.R., Slovak M.L., Arber D.A. (1999). Adult precursor-B acute lymphoblastic leukemia with translocations involving chromosome band 19p13 is associated with poor prognosis. Cancer Genet Cytogenet 109(l):58-65.
Khan J., Wei J.S., Ringnér M., Saal L.H., Ladanyi M., Westermann F., Berthold F., Schwab M., Antonescu C.R., Peterson C., Meltzer P.S. (2001). Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 7(6):673-9.
Li L., Pederson L.G., Darden T.A., Weinberg C.R. (2002). “Computational Analysis of Leukemia Microarray Expression Data Using the GA/KNN Method.” In Methods of Microarray Data Analysis, Lin S.M., Johnson K.F., eds., Norwell, MA, Kluwer Academic Publishers.
Mitchell T.M. (1997). Machine Learning. NY: McGraw-Hill Companies.
Olofsson A.M., Vestberg M., Herwald H., Rygaard J., David G., Arfors K.-E., Linde V., Flodgaard H., Dedio J., Muller-Esterl W., Lundgren-Åkerlund E. (1999). Heparin-binding protein targeted to mitochondrial compartments protects endothelial cells from apoptosis. J Clin Invest 104(7):885-94
Ostergaard E., Flodgaard H. (1992). A neutrophil-derived proteolytic inactive elastase homologue (hHBP) mediates reversible contraction of fibroblasts and endothelial cell monolayers and stimulates monocyte survival and thrombospondin secretion. J Leukoc Biol 51(4):316-23
Pearl J. (1988). Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. CA: Morgan Kaufmann Publishers.
Slonim D.K., Tamayo P., Mesirov J.P., Golub T.R., Lander E.S. (2000). Class prediction and discovery using gene expression data. Proc. of the Fourth Annual International Conference on Computational Molecular Biology; 2000 April 8 - 11; Tokyo. New York: ACM Press.
Spirtes P., Glymour C., Schemes R. (2000). Causation, Prediction, and Search, 2nd edition. MA: MIT Press.
Zhang B.-T., Hwang K.-B., Chang J.-H,, Augh S.J. Ensemble of Bayesian networks for gene expression-based classification of cancers. Artif Intell Med (submitted) 2002.
Chapter 9
CLASSIFYING MICROARRAY DATA USING SUPPORT VECTOR MACHINES
Sayan Mukherjee
PostDoctoral Fellow: MIT/Whitehead Institute for Genome Research and Center for Biological and Computational Learning at MIT,
e-mail: sayan@mit.edu
1.INTRODUCTION
Over the last few years the routine use of DNA microarrays has made possible the creation of large data sets of molecular information characterizing complex biological systems. Molecular classification approaches based on machine learning algorithms applied to DNA microarray data have been shown to have statistical and clinical relevance for a variety of tumor types: Leukemia (Golub et al., 1999), Lymphoma (Shipp et al., 2001), Brain cancer (Pomeroy et al., 2002), Lung cancer (Bhattacharjee et al., 2001) and the classification of multiple primary tumors (Ramaswamy et al., 2001).
One particular machine learning algorithm, Support Vector Machines (SVMs), has shown promise in a variety of biological classification tasks, including gene expression microarrays (Brown et al., 2000, Mukherjee et al., 1999). SVMs are powerful classification systems based on regularization techniques with excellent performance in many practical classification problems (Vapnik, 1998, Evgeniou et al., 2000).
This chapter serves as an introduction to the use of SVMs in analyzing DNA microarray data. An informal theoretical motivation of SVMs both from a geometric and algorithmic perspective, followed by an application to leukemia classification, is described in Section 2. The problem of gene selection is described in Section 3. Section 4 states some results on a variety of cancer morphology and treatment outcome problems. Multiclass classification is described in Section 5. Section 6 lists software sources and
9. Classifying Microarray Data Using Support Vector Machines |
167 |
rules of thumb that a user may find helpful. The chapter concludes with a brief discussion of SVMs with respect to some other algorithms.
2.SUPPORT VECTOR MACHINES
In binary microarray classification problems, we are given l experiments  This is called the training set, where
This is called the training set, where  is a vector corresponding to the expression measurements of the
is a vector corresponding to the expression measurements of the  experiment or sample (this vector has n components, each component is the expression measurement for a gene or EST) and
experiment or sample (this vector has n components, each component is the expression measurement for a gene or EST) and  is a binary class label, which will be
is a binary class label, which will be
± 1. We want to estimate a multivariate function from the training set that will accurately label a new sample, that is  This problem of learning a classification boundary given positive and negative examples is a particular case of the problem of approximating a multivariate function from sparse data. The problem of approximating a function from sparse data is illposed. Regularization is a classical approach to making the problem wellposed (Tikhonov and Arsenin, 1977).
This problem of learning a classification boundary given positive and negative examples is a particular case of the problem of approximating a multivariate function from sparse data. The problem of approximating a function from sparse data is illposed. Regularization is a classical approach to making the problem wellposed (Tikhonov and Arsenin, 1977).
A problem is well-posed if it has a solution, the solution is unique, and the solution is stable with respect to perturbations of the data (small perturbations of the data result in small perturbations of the solution). This last property is very important in the domain of analyzing microarray data where the number of variables, genes and ESTs measured, is much larger than the number of samples. In statistics when the number of variables is much larger that the number of samples one is said to be facing the curse of dimensionality and the function estimated may very accurately fit the samples in the training set but be very inaccurate in assigning the label of a new sample, this is referred to as over-fitting. The imposition of stability on the solution mitigates the above problem by making sure that the function is smooth so new samples similar to those in the training set will be labeled similarly, this is often referred to as the blessing of smoothness.
2.1Mathematical Background of SVMs
We start with a geometric intuition of SVMs and then give a more general mathematical formulation.
2.1.1A Geometrical Interpretation
A geometric interpretation of the SVM illustrates how this idea of smoothness or stability gives rise to a geometric quantity called the margin which is a measure of how well separated the two classes can be. We start by assuming that the classification function is linear

168 |
Chapter 9 |
where  and
and  are the
are the  elements of the vectors x and w, respectively. The operation
elements of the vectors x and w, respectively. The operation  is called a dot product. The label of a new point
is called a dot product. The label of a new point  is the sign of the above function,
is the sign of the above function, 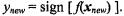 The classification boundary, all values of x for which f(x) = 0, is a hyperplane1 defined by its normal vector w (see Figure 9.1).
The classification boundary, all values of x for which f(x) = 0, is a hyperplane1 defined by its normal vector w (see Figure 9.1).
Assume we have points from two classes that can be separated by a hyperplane and x is the closest data point to the hyperplane, define  to be the closest point on the hyperplane to x. This is the closest point to x that satisfies
to be the closest point on the hyperplane to x. This is the closest point to x that satisfies  (see Figure 9.2). We then have the following two equations:
(see Figure 9.2). We then have the following two equations:
for some k, and
Subtracting these two equations, we obtain 
Dividing by the norm of w (the norm of w is the length of the vector w), we obtain:2
1 A hyperplane is the extension of the two or three dimensional concepts of lines and planes to higher dimensions. Note, that in an n-dimensional space, a hyperplane is an n – 1 dimensional object in the same way that a plane is a two dimensional object in a three dimensional space. This is why the normal or perpendicular to the hyperplane is always a vector.
2 The notation  refers to the absolute value of the variable a. The notation
refers to the absolute value of the variable a. The notation refers the length of the vector a.
refers the length of the vector a.

9. Classifying Microarray Data Using Support Vector Machines |
169 |
where  Noting that
Noting that  is a unit vector (a vector of length 1), and the vector
is a unit vector (a vector of length 1), and the vector is parallel to w, we conclude that
is parallel to w, we conclude that
Our objective is to maximize the distance between the hyperplane and the closest point, with the constraint that the points from the two classes fall on opposite sides of the hyperplane. The following optimization problem satisfies the objective:
Note that |
when the point x is the circle closest to the |
hyperplane in Figure 9.2. |
|
For technical reasons, the optimization problem stated above is not easy to solve. One difficulty is that if we find a solution w, then c w for any positive constant c is also a solution. This is because we have not fixed a scale or unit to the problem. So without any loss of generality, we will require that for the point  closest to the hyperplane k = 1. This fixes a scale and unit to the problem and results in a guarantee that
closest to the hyperplane k = 1. This fixes a scale and unit to the problem and results in a guarantee that  for all
for all  All other points are measured with respect to the closest point, which is distance 1 from the optimal hyperplane. Therefore, we may equivalently solve the problem
All other points are measured with respect to the closest point, which is distance 1 from the optimal hyperplane. Therefore, we may equivalently solve the problem

170 |
Chapter 9 |
An equivalent, but simpler problem (Vapnik, 1998) is
Note that so far, we have considered only hyperplanes that pass through the origin. In many applications, this restriction is unnecessary, and the standard separable (i.e. the hyperplane can separate the two classes) SVM problem is written as
where b is a free threshold parameter that translates the optimal hyperplane relative to the origin. The distance from the hyperplane to the closest points of the two classes is called the margin and is  SVMs find the hyperplane that maximize the margin. Figure 9.3 illustrates the advantage of a large margin.
SVMs find the hyperplane that maximize the margin. Figure 9.3 illustrates the advantage of a large margin.
In practice, data sets are often not linearly separable. To deal with this situation, we add slack variables that allow us to violate our original distance constraints. The problem becomes now:

9. Classifying Microarray Data Using Support Vector Machines |
171 |
where  for all i. This new formulation trades off the two goals of finding a hyperplane with large margin (minimizing
for all i. This new formulation trades off the two goals of finding a hyperplane with large margin (minimizing  and finding a hyperplane that separates the data well (minimizing the
and finding a hyperplane that separates the data well (minimizing the  The parameter C controls this trade-off. This formulation is called the soft margin SVM. The parameter C controls this trade-off. It is no longer simple to interpret the final solution of the SVM problem geometrically. Figure 9.4 illustrates the soft margin SVM.
The parameter C controls this trade-off. This formulation is called the soft margin SVM. The parameter C controls this trade-off. It is no longer simple to interpret the final solution of the SVM problem geometrically. Figure 9.4 illustrates the soft margin SVM.
SVMs can also be used to construct nonlinear separating surfaces. The basic idea here is to nonlinearly map the data to a feature space of high or possibly infinite dimensions,  We then apply the linear SVM algorithm in this feature space. A linear separating hyperplane in the feature space corresponds to a nonlinear surface in the original space. We can now rewrite Equation 9.6 using the data points mapped into the feature space, and we obtain Equation 9.7.
We then apply the linear SVM algorithm in this feature space. A linear separating hyperplane in the feature space corresponds to a nonlinear surface in the original space. We can now rewrite Equation 9.6 using the data points mapped into the feature space, and we obtain Equation 9.7.

172 |
Chapter 9 |
 for all i, where the vector w has the same dimensionality as the feature space and can be thought of as the normal of a hyperplane in the feature space. The solution to the above optimization problem has the form
for all i, where the vector w has the same dimensionality as the feature space and can be thought of as the normal of a hyperplane in the feature space. The solution to the above optimization problem has the form
since the normal to the hyperplane can be written as a linear combination of the training points in the feature space,
For both the optimization problem (Equation 9.7) and the solution (Equation 9.8), the dot product of two points in the feature spaces needs to be computed. This dot product can be computed without explicitly mapping the points into feature space by a kernel function, which can be defined as the dot product for two points in the feature space:
So our solution to the optimization problem has now the form:
Most of the coefficients  will be zero; only the coefficients of the points closest to the maximum margin hyperplane in the feature space will have nonzero coefficients. These points are called the support vectors. Figure 9.5 illustrates a nonlinear decision boundary and the idea of support vectors.
will be zero; only the coefficients of the points closest to the maximum margin hyperplane in the feature space will have nonzero coefficients. These points are called the support vectors. Figure 9.5 illustrates a nonlinear decision boundary and the idea of support vectors.

9. Classifying Microarray Data Using Support Vector Machines |
173 |
The following example illustrates the connection between the mapping into a feature space and the kernel function. Assume that we measure the expression levels of two genes, TrkC and SonicHedghog (SH), For each sample, we have the expression vector  We use the following mapping
We use the following mapping 
If we have two samples x and  then we obtain:
then we obtain:
which is called a second order polynomial kernel. Note, that this kernel uses information about both expression levels of individual genes and also expression levels of pairs of genes. This can be interpreted as a model that incorporates co-regulation information. Assume that we measure the expression levels of 7,000 genes. The feature space that the second order polynomial would map into would have approximately 50 million elements, so it is advantageous that one does not have to explicitly construct this map.
The following two kernels, the polynomial and Gaussian kernel, are commonly used:
2.1.2A Theoretical Motivation for SVMs and Regularization
SVMs can also be formulated as an algorithm that finds a function f that minimizes the following functional3 (note that this functional is basically Equation 9.7, rewritten in a more general way):
where the first term, the hinge loss function, is used to measure the error between our estimate and
and and
and  The expression
The expression  is a measure of smoothness, and the margin is
is a measure of smoothness, and the margin is  , The variable l is the number of training examples, and 1 / C is the regularization parameter that trades off between smoothness and errors. The first term ensures that the estimated function has a small error on the training samples and the second term ensures that this function is also smooth. This functional is a particular
, The variable l is the number of training examples, and 1 / C is the regularization parameter that trades off between smoothness and errors. The first term ensures that the estimated function has a small error on the training samples and the second term ensures that this function is also smooth. This functional is a particular
3 A functional is a function that maps other functions to real numbers.