

Раздаточные материалы - 2003 / solid
.docЭЛЕКТРОННАЯ СТРУКТУРА ТВЕРДЫХ ТЕЛ
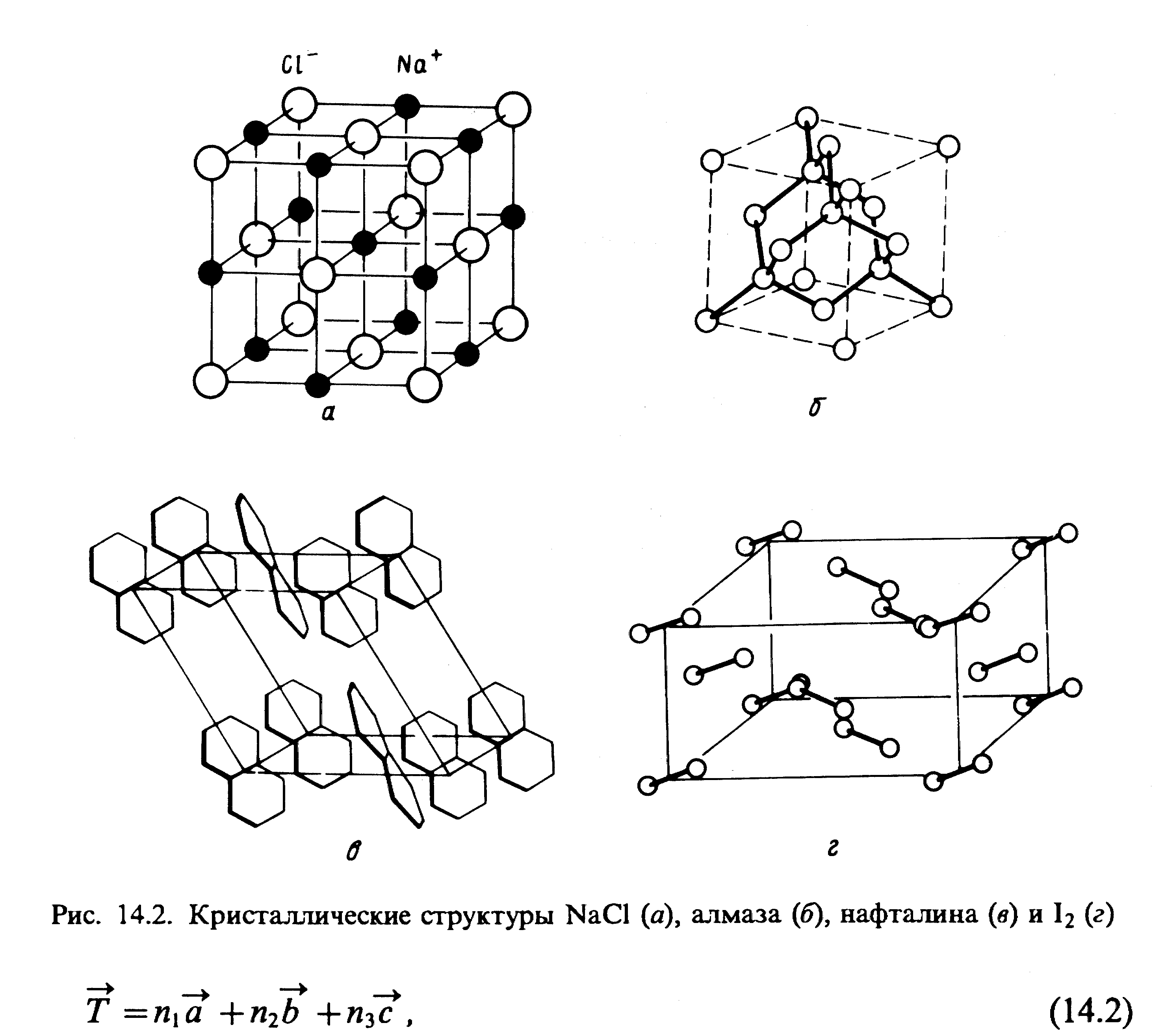
Твердое тело можно рассматривать как очень большую молекулу, в которой соблюдается либо дальний (идеальный), либо ближний порядок расположения атомов или молекул. Первый случай отвечает идеально регулярным кристаллам, симметрия которых описывается 230 федоровскими пространственными группами. Второй – более общий - случай охватывает реальные кристаллы с дефектами кристаллической решетки, аморфные тела, полимеры и т.д.: для них характерна лишь локальная симметрия определенных фрагментов. Для каждого из упомянутых случаев существуют свои способы описания электронной структуры. Идеально регулярные кристаллы считаются бесконечными (наличие поверхности игнорируется) и их волновые функции определяются с явным учетом трансляционной симметрии кристалла. В кристаллах с ближним порядком часто достаточно использовать модифицированные молекулярные модели, выделив некоторый атомный или молекулярный фрагмент – кластер. Рассмотрим оба этих подхода подробнее.
Одноэлектронные волновые функции в бесконечных периодических кристаллах и методы их расчета
Симметрийные свойства идеальных кристаллов состоят в следующем.
В идеальном кристалле всегда можно ввести три вектора трансляций: a,b и c так, что физические свойства кристалла в некоторой произвольно выбранной точке r точно воспроизводятся в любой другой точке r удовлетворяющей условию
![]() ( 1
)
( 1
)
где n1, n2, n3 —произвольные целые числа. Совокупность точек r, определяемая выражением (1), при различных n1, n2, n3 дает кристаллическую решетку, которая описывает регулярное периодическое расположение точек в пространстве.
Параллелепипед, имеющий в качестве ребер вектора : a,b и c , называется
элементарной ячейкой ( рис. 1). Перемещение всего кристалла как целого параллельно самому себе, описываемое вектором Т, называется трансляцией. Вектор трансляции кристаллической решетки связывает любые две точки ре- шетки.
Посредством соответствующих операций трансляций элементарной ячейкой можно заполнить все пространство кристаллической структуры. Такое свойство кристалла названо трансляционной симметрией. На рис. 2 представлены структуры некоторых атомных и молекулярных кристаллов.
Трансляционная симметрия предполагает бесконечную протяженность кристалла. Конечно, регулярные структуры не являются бесконечными, а при отсутствии бесконечности теряется важное свойство трансляционной симметрии.
Один из способов сохранения трансляционной симметрии конечных систем - наложение циклических граничных условий (условия Борна-фон-Кармана). Суть их в том, что эквивалентные группы атомов отождествляются друг с другом так, чтобы граница отсутствовала.
В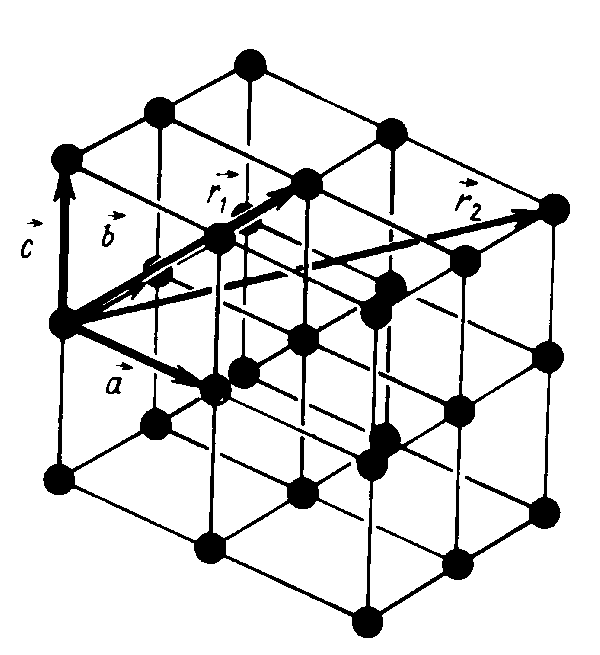 ажнейшие
электронные свойства периодических
кристаллов хорошо видны уже в случае
одномерных систем. Для цепочки атомов
это это равносильно изгибу цепочки в
окружность большого радиуса и соединение
между собой концевых атомов. Рассмотрим
цепочку, состоящую из атомов, разделенных
расстоянием а
и имеющих один валентный электрон (
Li, Na, Ka и
т.д.), описываемый атомной орбиталью
(х).
Каждое ядро в этих атомах хорошо
экранировано внутренними электронами
и валентный электрон лишь слабо связан
со «своим» остовом. Влияние соседних
атомом в цепочке приводит к тому, что
валентные электроны могут передвигаться
от атома к атому, как если бы они были
свободными (модель почти свободных
электронов Зоммерфельда). Волновая
функция свободного электрона есть
бегущая волна exp(ikx),
а его кинетическая энергия равна
ажнейшие
электронные свойства периодических
кристаллов хорошо видны уже в случае
одномерных систем. Для цепочки атомов
это это равносильно изгибу цепочки в
окружность большого радиуса и соединение
между собой концевых атомов. Рассмотрим
цепочку, состоящую из атомов, разделенных
расстоянием а
и имеющих один валентный электрон (
Li, Na, Ka и
т.д.), описываемый атомной орбиталью
(х).
Каждое ядро в этих атомах хорошо
экранировано внутренними электронами
и валентный электрон лишь слабо связан
со «своим» остовом. Влияние соседних
атомом в цепочке приводит к тому, что
валентные электроны могут передвигаться
от атома к атому, как если бы они были
свободными (модель почти свободных
электронов Зоммерфельда). Волновая
функция свободного электрона есть
бегущая волна exp(ikx),
а его кинетическая энергия равна
Екин= p2/2m=(kh)2/82m ( 2)
(k=2/ - волновое число или квазиимпульс электрона). Специфика нашей задачи состоит в том, что движение электрона происходит в слабом периодическом потенциале, вид которого обусловлен сортом и расположением атомов в решетке. Это означает, что потенциал V(x) в уравнении Шредингера
[T(x) + V(x)] uk(x) = Euk(x) ( 3 )
обладает свойством V(x)=V(x+na), где n=0,1, 2, … Учет этого обстоятельства при решении уравнения Шредингера приводит к тому, что волновая функция электрона в периодическом потенциале uk(x) имеет вид модулированной зависящей от волнового числа k бегущей волны
uk(x)=N n exp(ikan)( x + na) ( 4 )
(N – нормировочный множитель), которая также периодична в решетке: (x) = (x+na) и сдвиг цепочки на величину, кратную а, не меняет uk. Функция uk, играющая в твердом теле роль атомной орбитали, называется функцией Блоха. При k = b/a волновые функции электрона уже не являются бегущими волнами типа exp( ikx). Так, при b=0 k=0 и
u0(x)=N [( x )+( x + a)+ (x + 2a)+ (x + 3a) +…] , ( 5 )
а при b=1 k= /a и
u/a (x)=N [( x )-( x + a)+ (x + 2a)- (x + 3a) +…] (6 )
( см. рис.3) . В первом случае АО на разных атомах интерферируют в фазе и результирующая функция Блоха является связывающей и отвечает более низкой энергии. Второй случай отвечает деструктивной интерференции АО и,
a) k = 0 u0 = N [ (x) + (x + a) + ....]
![]()
б) k = + /a u/a = N [ (a) - (x + a) + .....]
![]()
Рис.3
соответственно, - разрыхляющей функции Блоха. Таким образом, волновые функции электрона в периодическом потенциале можно ассоциировать со стоячими волнами вида u1=sinx/a и u2 =cosx/a , каждая из которых есть сумма бегущих волн (рис. 4). Эти стоячие волны имеют узлы и пучности в разных областях пространства по отношению к создающим внешний потенциал атомным остовам. Это значит, что они отвечают концентрации электронов (которая пропорциональна u2) в местах, отвечающих различным значениям потенциальной энергии. Так, u2 описывает концентрации электронов вблизи остовов, где потенциальная энергия минимальна, а u1 - между остовами.
Число возможных значений k определяется числом узлов кристаллической решетки. Область в k –пространстве, лежащая между /a, называется первой зоной Бриллюэна. Поведение электронов в более высоких зонах Бриллюэна
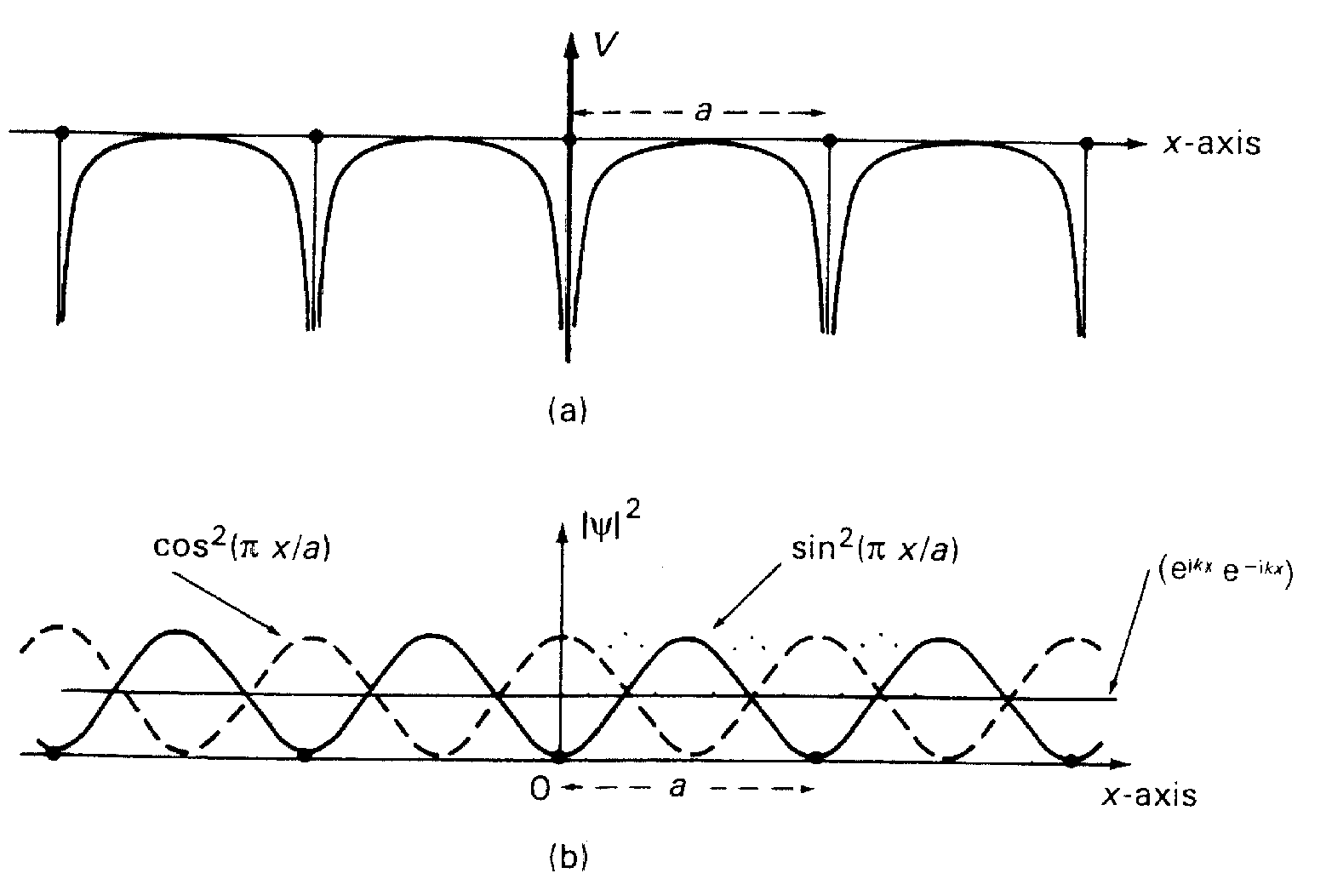 Рис.4
Рис.4
получено из информации о первой зоне (рис.5). В пределах первой зоны Бриллюэна энергия по-прежнему имеет вид, близкий к (2). Однако на границах зон Бриллюэна среди возможных (“разрешенных”) значений энергии появляются разрывы из-за того, что средняя потенциальная энергия электрона, описываемого функцией u2 меньше, чем для случая u1. Разность энергий Eg называется энергетической щелью.
рис.5
Кристаллические орбитали – аналоги МО – строятся из функций Блоха (4), которые таким образом играют в кристалле роль базисных функций:
k (r ) = cj(k) ukj(r) (7)
Т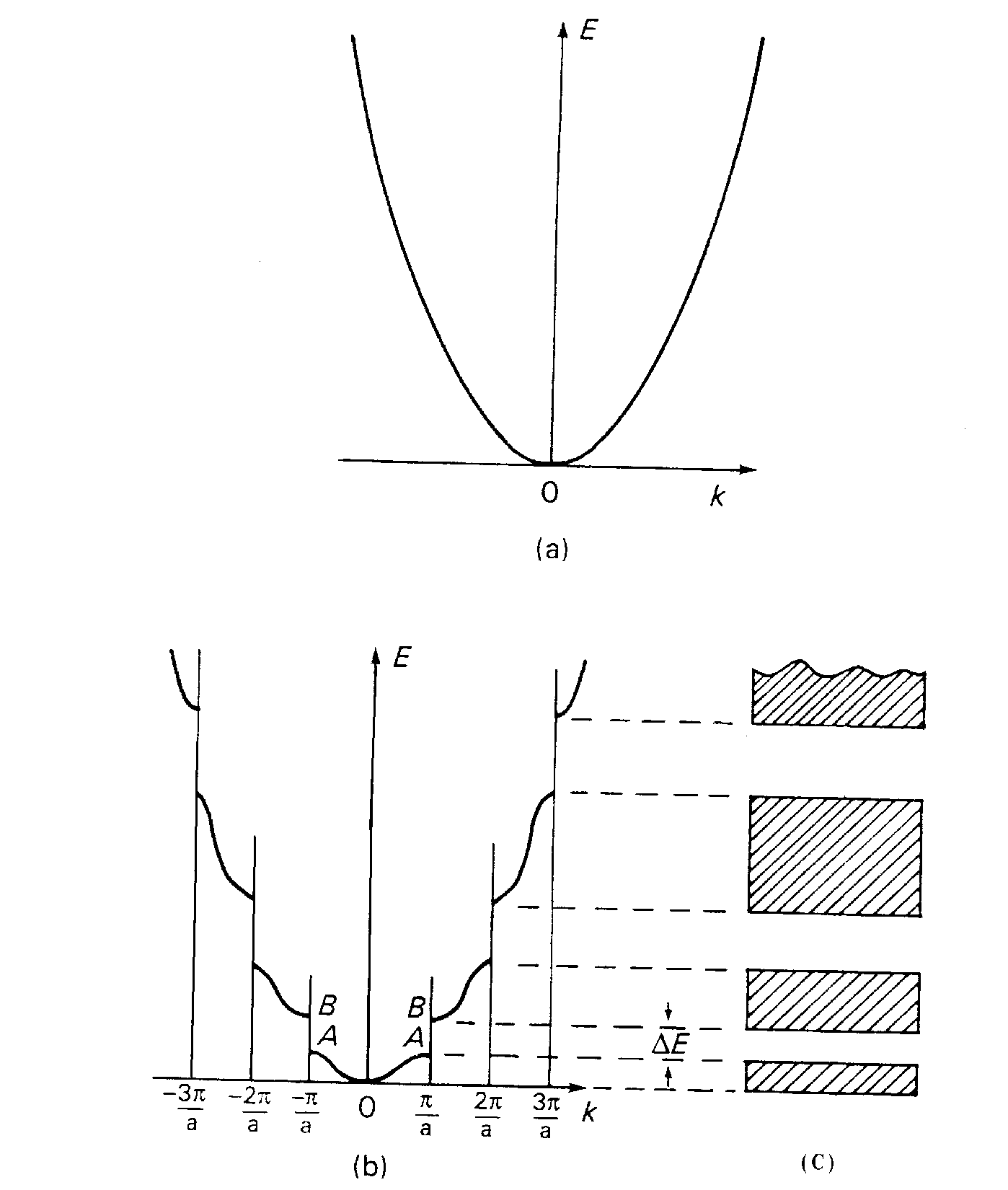 аким
образом, электроны в кристалле оказываются
распределенными по энергетическим
полосам (зонам), состоящим из уровней
энергии, отвечающих волнам с разрешенными
симметрией волновыми векторами.
Расстояния между энергетическими
уровнями на краях зоны меньше, чем в
середине, т. е. плотность уровней (число
уровней на единицу энергии) не одинакова
по всей зоне: у краев
зоны
плотность выше. Для описания зависимости
расстояния между
уровнями
от
k
предполагают
непрерывное
заполнение
зоны (полосы)
энергетическими уровнями
и вводят
непрерывную функцию Е(k),
которую называют также структурой
полосы. Она
описывает
закон дисперсии энергии.
Высшая орбиталь в твердом теле, заполненная
электронами, называется уровнем
Ферми.
аким
образом, электроны в кристалле оказываются
распределенными по энергетическим
полосам (зонам), состоящим из уровней
энергии, отвечающих волнам с разрешенными
симметрией волновыми векторами.
Расстояния между энергетическими
уровнями на краях зоны меньше, чем в
середине, т. е. плотность уровней (число
уровней на единицу энергии) не одинакова
по всей зоне: у краев
зоны
плотность выше. Для описания зависимости
расстояния между
уровнями
от
k
предполагают
непрерывное
заполнение
зоны (полосы)
энергетическими уровнями
и вводят
непрерывную функцию Е(k),
которую называют также структурой
полосы. Она
описывает
закон дисперсии энергии.
Высшая орбиталь в твердом теле, заполненная
электронами, называется уровнем
Ферми.
Распределение энергетических уровней в зоне характеризует плотность состояний:
D(E) = dN(E)/dE (8)
Интегрирование D(E) по dE до уровня Ферми дает полное число кристаллических орбиталей.
Разность между высшим и низшим энергетическими уровнями называют шириной зоны (полосы), она также характеризует дисперсию энергии. Ширина зоны определяется величиной взаимодействия между атомами в соседних ячейках и зависит от соответствующих интегралов перекрывания АО.
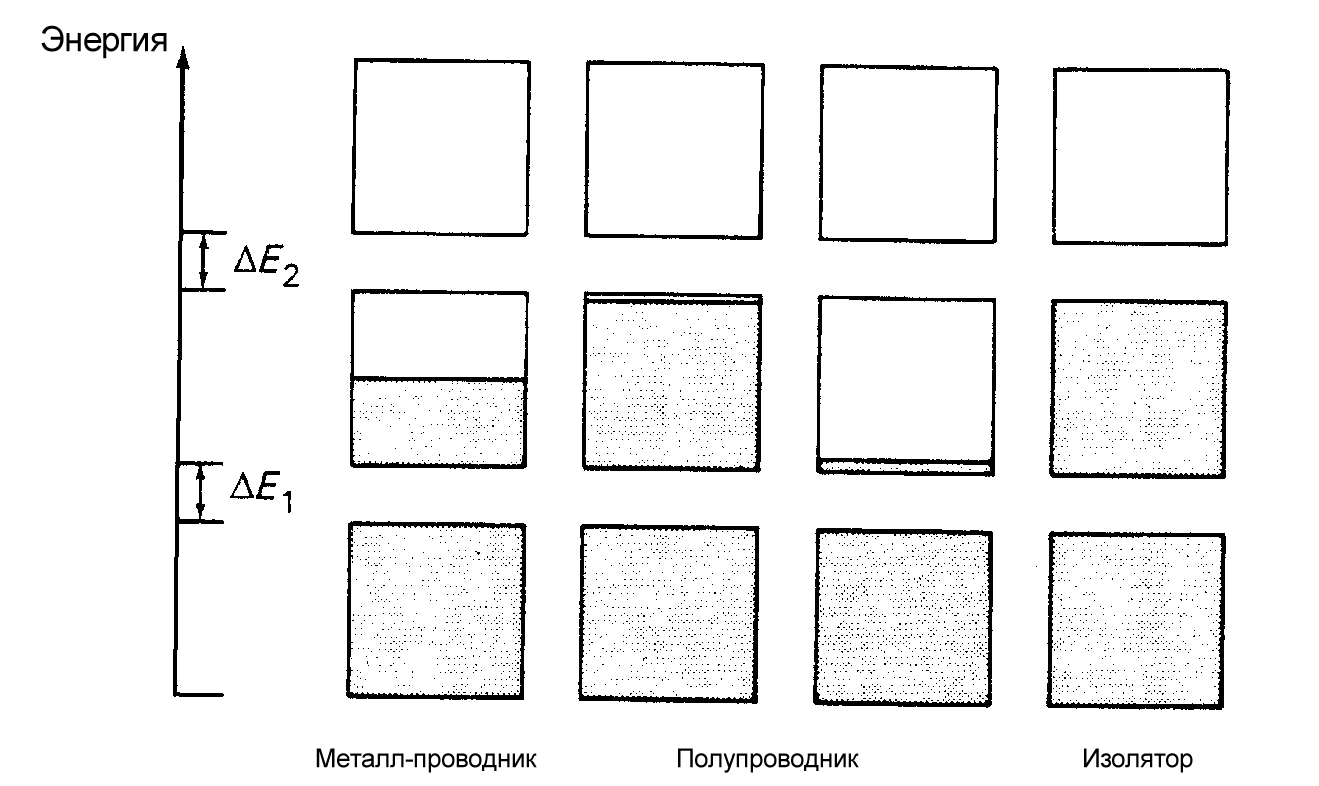
Промежуток, между верхней энергетической зоной, заполненной элект-ронами (валентной), и нижней, не заполненной электронами (зоной проводи-мости) называется запрещенной зоной.
От того, как заполнена валентная зона и какова ширина запрещенной зоны, зависят многие свойства веществ. Зона Бриллюэна содержит столько разрешенных значений k- векторов ( а значит, столько различных волновых функций), сколько элементарных ячеек N в кристалле. Если кристалл образовался из
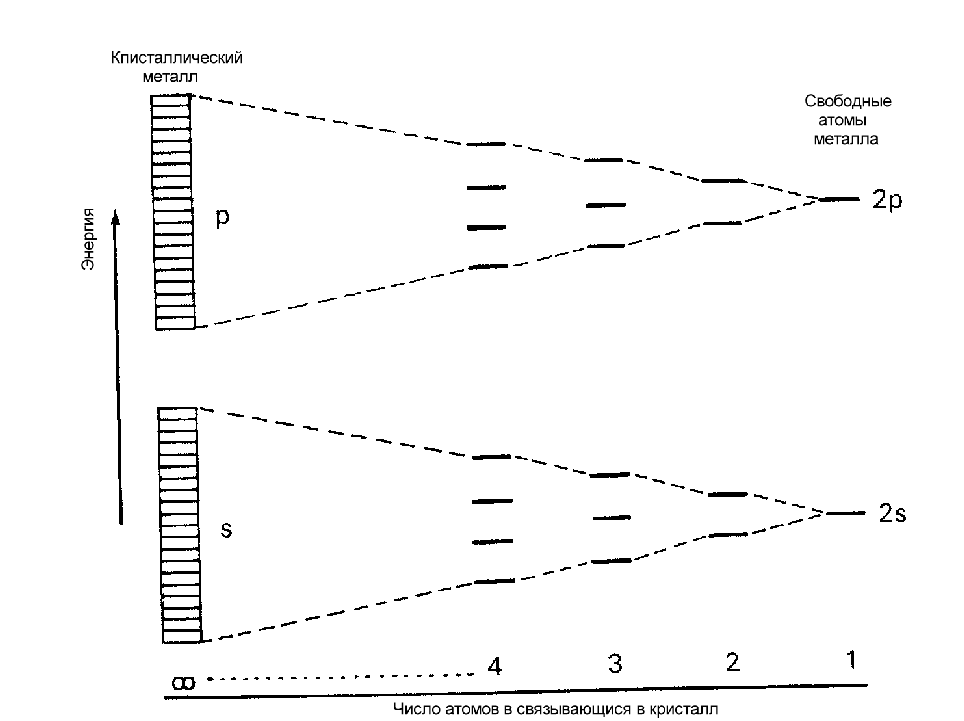
атомов с g валентными электронами (g=1 для s оболочки; g=3 для p оболочки и т.д.), то зона состоит из Ng энергетических уровней, каждый из которых может
быть занят 1 или 2 электронами (рис.6). Если валентная зона полностью запол-нена электронами, а ширина запрещенной зоны Еg 2 эВ, то вещество называется диэлектриком. Если валентная зона полностью заполнена электронами, а ширина запрещенной зоны Еg 2 эВ, то вещество называется полупроводником.
Если валентная зона заполнена электронами частично либо перекрывается с зоной проводимости, то вещество называется металлом.
Для расчета одноэлектронных волновых функций в кристаллах используют два основных метода.
-
Метод Хартри-Фока. Функции (4) используются как базисные и строятся из атомных орбиталей ( как правиоло, ОСТ). Недостатки: из-за учета только корреляции Паули переоценивает ширину запрещенной зонв в 1,5-2 раза; неприменим для расчета Ферми-поверхности металлов.
-
Метод Кона-Шэма – относится к группе методов функционала плотности.
Основан на предположении, что электронную плотность можно рассматривать как неоднородный электронный газ, определяющий обменно-корреляционные эффекты. Одноэлектронные уравнения имеют вид ( атомные единицы)
[ -1/22 + VN(r) +(r)/(r- r)dr +Vxc ] k (r ) = E(k)k (r ) (9)
Здесь VN –потенциал ядер, Vxc – обменно-корреляционный потенциал,для которого используются различные приближения – функционалы ЭП. Базисные функции различны: это могут быть как комбинации атомных орбиталей, так и наборы плоских волн. Недостаток: недооценка ширины запрещенной зоны.
Кластерные методы
Часто ( например, при расчетах нестехиометрических кристаллов) целесообразно рассчитать лишь некоторый атомный или молекулярный фрагмент кристалла – кластер. Основной метод, используемый для это цели– метод Хартри-Фока.
Одна из распространенных его кластерных модификаций – так называемый X-метод. Корреляция электронов здесь по-прежнему игнорируется, а обменный потенциал представляется в виде
Vx(r ) = -3((3/8)(r ))1/3 (10)
Несмотря на кажущуюся переупрощенность, X-метод дает хорошее количественное описание не только локализованных состояний, но и зонной структуры твердых тел. Для этого используют метод «погруженного» кластера: одноэлектронные функции раскладывают по базису из локализованных орбиталей и функций Блоха. Метод успешно применяется для изучения магнитных свойств веществ, химической связи и др.