

Multiple Bonds Between Metal Atoms / 16-Physical, Spectroscopic and Theoretical Results
.pdf16
Physical, Spectroscopic and
Theoretical Results
F. Albert Cotton,
Texas A&M University
16.1 Structural Correlations
16.1.1 Bond orders and bond lengths
Throughout the preceding chapters of this book we have reported and in some cases discussed the lengths of the multiple bonds between metal atoms. We have reported in tabular form most of the M–M distances that have been determined crystallographically. It is now time to make some general comments on these distances in relation to our understanding of the M–M bonds.
It is a general qualitative rule in chemistry that bond lengths and bond orders are inversely related. In some limited areas, particularly with the first-row elements carbon, nitrogen, and oxygen, quantitative relationships expressing bond length as a single-valued function of bond order are well known. However, these quantitative relationships are based heavily on faith, as well as fact. Bond lengths are facts; bond orders are not. In the process of inferring a bond order from a bond length one is often getting out no more than what was put in to begin with.
We shall not further digress into a discussion of the philosophical ambiguities of these quantitative bond order-bond length correlations because they have proven to be useful, within their own sphere of application. However, there is no a priori reason to expect that similar procedures will (or will not!) work in the very different realm of metal-to-metal bonds. Experience is the only test, and experience thus far has shown that M–M bonds cannot usefully be treated in such a way.
In the realm of M–M bonds it is best to invoke only a qualitative inverse relationship between bond order and bond length and to define bond order only in a qualitative or ordinal way. In this book we have used the term M–M bond order only to indicate how many electron pairs are believed, on the basis of essentially qualitative considerations, to play a significant part in holding the pair of metal atoms together. We condemn as foolish and hopeless any effort to associate a unique, precise, quantitative bond order with each and every M–M internuclear distance.
In principle, M–M bond lengths, like others, should be amenable to treatment by the method of molecular mechanics, and some efforts1-4 (hampered by a dearth of necessary experimental data) to do this have been reported. In general the results are encouraging and can account for
707

708Multiple Bonds Between Metal Atoms Chapter 16
variations in the lengths and vibrational frequencies of a given type (i.e. order) of M–M bond by taking into consideration the way in which the entire set of intra-ligand bonds and ligandligand repulsions contribute to the net result. One major result of this work has been to support and clarify the concept that bridging ligands (RCO2- and stereoelectronically similar ones) favor shorter M–M distances (see Section 4.4.5 for Mo–Mo bonds) and high M–M stretching frequencies. Such molecular mechanics calculations have also provided some estimates of the barriers to internal rotation that arise from a combination of the β-bonding and the end-to-end ligand–ligand repulsive forces (see Section 16.1.6).
Another point worth noting here is that even the statement just made concerning “how many electron pairs... play a significant part in holding the metal atoms together” has a simplicity that is very deceptive. The classic molecular orbital definition of bond order, as an ordinal number, is valid only when the bond is described by a single electron configuration. But, as we have already noted for Cr–Cr bonds in Chapter 3, and will discuss more generally later, multiple bonds between metal atoms can rarely if ever be described accurately without including configuration interaction (CI). This means that the best description of the ground state (or any other state, for that matter) entails the mixing in of configurations having fewer bonding electrons and more antibonding electrons. Thus the net value of (nb - na)/2 (i.e. bonding pairs minus antibonding pairs) in general is a fractional number, with a value that keeps changing as the CI calculation is extended.
Thus, when we have referred to double bonds, triple bonds, and quadruple bonds, i.e. bond orders of 2, 3, and 4, we have been using somewhat fictional or formal numbers, often corresponding to what we would deduce if we carried out a simple (i.e. one-configuration) HartreeFock calculation, then used the results to calculate an integral or half-integral bond order, and ignored CI. Despite the reservations we must have about this procedure, the bond orders so obtained provided a much-needed framework for classifying and discussing the subject.
Even at the most empirical level both the shortcomings and the utility of the nominal bond orders are apparent. For example Mo–Mo quadruple bond distances range from about 2.07 Å to about 2.18 Å, as noted in Section 4.5.5. There is a range of values for Mo–Mo triple bonds as well, running from 2.167 Å in Mo2(CH2SiMe3)6 to 2.222(2) Å in Mo2(OCH2CMe3)6. If one were to insist that every different Mo–Mo distance must be associated with a different bond order, there would be, in addition to the irksome difficulty of devising a meaningful way of establishing the proper numerical values, the insuperable inconsistency of having to assign a higher bond order in the triply bonded Mo2(CH2SiMe3)6 than in the quadruply bonded [Mo2(NCS)8]4- ion.
Deeper insight into some of the reasons why bond order and bond length seldom have a truly simple relationship (even if there is sometimes an apparently simple one) is provided by the following story.5 The following data for an essentially isostructural series of compounds appear to provide a straightforward example of bond length increasing simply as a function of decreasing bond order:
|
Bond length (Å) |
Elect. conf. |
Bond order |
[Mo2(SO4)4]4- |
2.111(1) |
μ2/4β2 |
4.0 |
[Mo2(SO4)4]3- |
2.167(1) |
μ2/4β |
3.5 |
[Mo2(HPO4)4]2- |
2.223(2) |
μ2/4 |
3.0 |
From this it would seem to follow straightforwardly that on oxidizing the [Tc2Cl8]3- ion, which has a μ2/4β2β* configuration, to [Tc2Cl8]2-, the loss of the β* electron should strengthen and shorten the bond, by something of the order of 0.05-0.10 Å. The actual results, shown below are opposite to this expectation.

Physical, Spectroscopic and Theoretical Results 709
|
|
|
Cotton |
|
Bond length (Å) |
Elect. conf. |
Bond order |
[Tc2Cl8]3- |
2.105(1), 2.117(2) |
μ2/4β2β* |
3.5 |
[Tc2Cl8]2- |
2.151(1) |
μ2/4β2 |
4.0 |
The reason for this initially surprising result is as follows. First, we must recognize that the μ and / components of these bonds are far stronger than the β (or β*) component, the latter supplying only a few per cent of the total bond strength. Therefore even a small change (2-3%) in the μ and / contributions will be as important as a 50% change in the β-bonding. Second, in all of the five compounds just discussed, the changes in bond order (by removing β or β* electrons) are also accompanied by increases in the oxidation states of the metal atoms. An increase in the effective positive charge on the metal atoms may be expected to cause some contraction of the d-orbitals and hence a diminution of the overlaps in all components of the M–M bonds, including the strong μ and / components.
In the case of the three molybdenum compounds, there is an increase in formal positive charge along with the reduction in bond order. Thus, both factors tend to increase the M–M distance, and increases are, therefore, necessarily observed. It is a mistake, however, to attribute them entirely to the reduction in bond order. In the case of the two technetium compounds, however, the two factors work in opposite directions, and evidently the bond-weakening effect of increased effective charge on the metal atoms outweighs the bond-strengthening effect of removing the β* electron. A net increase in Tc–Tc distance thus results despite the increase in bond order.
While the line of argument just developed might be regarded as simply ad hoc on the basis of only two cases, convincing support has been provided by the data for the following series of compounds,6 as well as others.
|
Bond length (Å) |
Elect. conf. |
Bond order |
Re2Cl6(PEt3)2 |
2.222(3) |
μ2/4β2 |
4.0 |
Re2Cl4(PEt3)4 |
2.232(6) |
μ2/4β2β*2 |
3.0 |
Re2Cl4(PMe2Ph)4 |
2.241(1) |
μ2/4β2β*2 |
3.0 |
[Re2Cl4(PMe2Ph)4]+ |
2.218(1) |
μ2/4β2β* |
3.5 |
[Re2Cl4(PMe2Ph)4]2+ |
2.215(2) |
μ2/4β2 |
4.0 |
The first two compounds show a possible slight lengthening with decreasing bond order, but evidently the charge factor, while not outweighing the bond order factor, is of approximately equal importance. An even better illustration of these effects is provided by the last three species, where there is no change in composition. The steady increase in bond order produces only a small and irregular decrease in bond length as a result of the countervailing effect of the steady increase in effective charge on the metal atoms.
Further support for the foregoing analysis is provided by the results of several kinds of experiments in which the bond order is changed without changing the formal oxidation states of the metal atoms. Two ways to do this, both of which will be discussed in detail later, are:
1.to reduce the β-bond strength by twisting the rotational conformation away from eclipsed towards the staggered, and
2.to excite an electron from a bonding to an antibonding orbital, as for example, in a βΑβ* type transition.
The very disparate importance of / (or /*) and β (or β*) electrons is clearly illustrated by
two ruthenium compounds. |
|
|
|
Bond length (Å) |
Elect. conf. |
Ru2(mhp)4 |
2.235(1) |
μ2/4β2/*2β*2 |
Ru2(PhN3Ph)4 |
2.399(1) |
μ2/4β2/*4 |
710Multiple Bonds Between Metal Atoms Chapter 16
Here there is no change in bond order, but two β* electrons are replaced by two /* electrons. Since the former are only weakly antibonding and the latter strongly so, a large increase in bond length would be expected, and is found.
Finally, we note that the very short Tc–Tc distance in K2Tc2Cl6, 2.04 Å, is, in fact, consistent with other Tc–Tc distances, such as 2.15 Å in [Tc2Cl8]2- and 2.11 Å in [Tc2Cl8]3-, because we now have [Tc2Cl8]4- ions fused together in chains. Within each such unit the low formal charge (Tc24+) allows strong μ and / overlap even though there are two β*-electrons.
16.1.2 Internal rotation
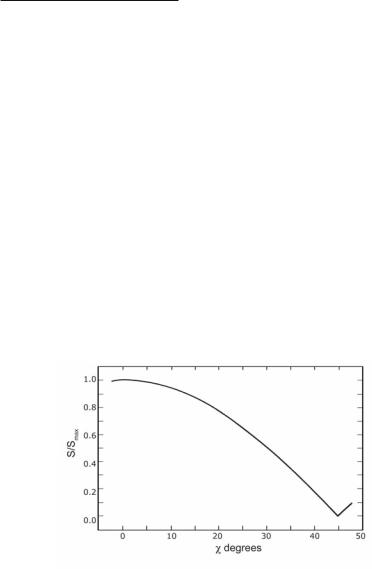
The strength of the μ and / components of multiple M–M bonds is essentially independent of the angle of internal rotation, since the μ overlap and the two / overlaps jointly are cylindrically symmetrical. Thus, in bonds of order 3 there is no inherently preferred angle of internal rotation, and the angle adopted is not prejudiced by the M–M bonding. In quadruple bonds (and to a lesser extent those of order 3.5) the presence of β bonding introduces an additional factor. The β overlap is angle-sensitive in a way that is easily determined7 from the angular wave function of the dxy-orbitals, with the M–M direction as the z axis. If we define the angle of internal rotation, ρ, as zero for the eclipsed structure and 45˚ for the perfectly staggered structure (symmetries of D4h and D4d for an M2X8 species), the overlap (S) varies as cos2ρ. A plot of Sβ versus ρ is shown in Fig. 16.1. Naturally, a plot of Sβ versus cos2ρ should be a straight line from Sβ = 1 (at ρ = 0) to Sβ = 0 (at ρ = 45˚).
Fig. 16.1. A plot of the β overlap vs torsion angle ρ.
The fact that the strength of the β-bond is greatest for ρ = 0 does not necessarily mean that this is the preferred angle when all factors are taken into account, however. The β bond is a relatively weak one, and the minimization of nonbonded repulsions, operating between the sets of ligands at the two ends of an L4M ML4 unit, may favor a rotation away from the eclipsed (ρ = 0) conformation. The value of ρ where these two forces are balanced may be expected, in general, to be other than zero. There are, of course, still other factors bearing on the result, including intermolecular (packing) forces. When some of the ligands are bidentate ones, such as Ph2PCH2CH2PPh2, that span the two metal atoms, the conformational preferences of the resulting M2-containing rings often favor twist angles quite different from zero. It is precisely this phenomenon that has allowed us to obtain the data required to map quantitatively the β manifold, as explained in Section 16.4.1.
Physical, Spectroscopic and Theoretical Results 711
Cotton
From Fig. 16.1 it can be seen that small values of ρav cause little diminution in the strength of the β-bond. A rotation of 22.5˚ (halfway toward a staggered conformation) costs only 30% of the β overlap, and even a 30˚ rotation leaves 50% of the β overlap intact. Thus, it is to be expected that in most cases when a quadruply bonded species is unconstrained by its surroundings, the optimum angle may well be > 0.
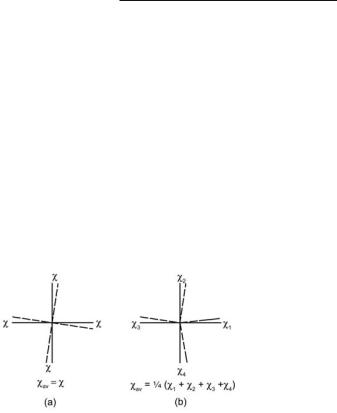
In some experimentally determined structures a C4 axis makes all four torsion angles equal, Fig. 16.2(a). In general, however, we require a practical rule for specifying the torsion angle. The rule that has been normally used is to define the four torsion angles as shown in Fig. 16.2(b) and use their average value (algebraic sum divided by 4). There are many examples where symmetry considerations result in the torsion angle (sometimes also called the twist angle) being rigorously zero when so defined. Thus, if there is a center of inversion at the midpoint of the M–M bond, the four torsion angles must form two pairs each of the same value but opposite in sign. This is a rather common occurrence but there are also other cases when the average of individually non-zero torsion angles is zero because of a plane or C2 axis of symmetry.
Fig. 16.2. Diagrams defining the angle of internal rotation for L4M–ML4 systems.
(a) The case where four fold symmetry prevails and all four L–M–M–L torsion angles are equal; (b) a general case where there is no overall symmetry and all four torsion angles may have different magnitudes and directions. Directions are defined consistently, so that if ρ1 and ρ4 are + or −, ρ2 and ρ3 are − or +.
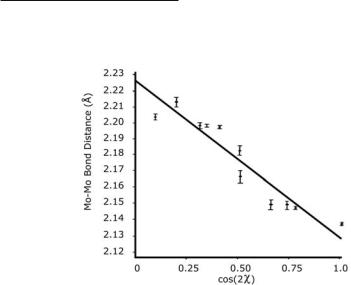
If bond length is linearly related (inversely) to the magnitude of Sβ, then for a series of compounds in which only the twist angle is changed, a plot of the M−M distance versus cos2ρ should be linear. Compounds of the type `-Mo2X4(PP)2, where X = Cl or Br and PP is a diphosphine, are a series that can be used to test this proposal.8,9 The results are shown in Fig. 16.3. The best straight line, shown in the figure, has a correlation coefficient of 0.995, and from it a change in Mo–Mo distance of 0.097 Å for complete loss of the β-bond (change in ρ from 0 to 45˚) is derived.
To make a quantitative estimate of the barrier to internal rotation, either experimentally or theoretically, experimental efforts have been devoted to 1H NMR line-shape measurements on dimolybdenum and ditungsten porphyrin compounds.10-13 In all cases the results have been in the range of 10-13 kcal mol-1. The “barrier to rotation” measured in this way is the difference in free energy between the most stable rotational conformation (not necessarily of D4h symmetry but probably close) and the least stable one (not necessarily of D4d symmetry, but probably close). How much these barriers tell us about the strength of the β bonding per se is more problematical than one might suppose. To say that the measured barrier and the β bond strength are equal is not justified.
Conversely, from theoretical calculations of the difference between the electronic energies of the D4h and D4d ground states, it is problematic to infer the measurable barrier to rotation.
712Multiple Bonds Between Metal Atoms Chapter 16
Efforts to estimate this electronic energy difference (see Section 16.3.2) have been made for Re2Cl82- and for Mo2Cl4(PH3)4, with results in the neighborhood of 12 kcal mol-1.
Fig. 16.3. A plot of Mo–Mo distances versus cos2ρ for eleven `-Mo2X4(LL)2 compounds, where LL is a diphosphine.
More recently, DFT calculations have been reported on Mo2Cl4(PH3)4 type molecules, both the 1,3,6,8 and 1,2,7,8 isomers.14-16 While such calculations are less rigorous electronically, they have the advantage of allowing for structural relaxation. They support previous calculations of an electronic barrier of 10-13 kcal mol-1. An interesting new result is that for Mo2Cl4(PH3)4 the 1,2,7,8 (C2h) structure is about 27 kcal more stable than the 1,2,5,6 (C2v) rotamer.
16.1.3 Axial ligands
There are many cases where a multiply bonded M2X8 or paddlewheel species also has axial ligands (or, less commonly, one axial ligand). The question of how the formation of bonds to these axial ligands affects the length of the M–M bond is an important one. Some qualitative generalizations are possible, but the magnitude of the effect varies greatly from one metal and one class of compound to another. As a broad generalization, however, the presence of axial ligands causes elongation of the M–M bond. This is best demonstrated by cases where a given M2X8 species has been structurally characterized both with and without axial ligands.
We have seen in Chapter 3 that the lengths of Cr–Cr bonds are extremely sensitive to the presence of axial ligands, and to the number and basicity of such ligands. The dichromium compounds are exceptional, however, and most other M–M multiple bonds are less responsive to the attachment of axial ligands and bind them less strongly. Consider, for example, the marked contrast between dichromium and dimolybdenum species. For the molybdenum compounds,17,18 even when axial ligands are attached, they are very weakly bound and the Mo–Mo distance is lengthened by only a few hundredths of an Angstrom.
With dirhenium compounds there is also only a small tendency to bind axial ligands and only small consequences. A good illustration of this is provided by the structure19 of Cs2Re2Cl8·H2O, which contains one [Re2Cl8]2- ion with no axial water molecules, and r(Re–Re) = 2.237(2) Å, and another with axially coordinated water molecules where r(Re–Re) = 2.252(2) Å. The water molecules in the latter are very loosely held, with r(Re–O) = 2.66(3) Å. Of course, the tendency of [Re2Cl8]2- to attract additional ligands is probably reduced by the fact that it is an anion. An [Re2(O2CR)4]2+ unit, being a cation, tends to form stronger bonds to axial ligands such as

Physical, Spectroscopic and Theoretical Results 713
Cotton
Cl-, with Re–Cl distances of 2.477(3) Å in Re2(O2CCMe3)4Cl2,20 which may be compared with Re–Cl distances19 of about 2.32 Å in [Re2Cl8]2-.
An interesting assessment of the effect of axial ligation in weakening M–M quadruple bonds was derived from the vibrational spectra of the cisoid M2(O2CCH3)2Cl4L2 compounds, where M = Tc, Re and L was varied through the series H2O, DMF, dimethylacetamide, DMSO, Ph3PO and pyridine.21 As the donor strength of L increased (in the above order), the metal– metal stretching frequencies decreased from 311 cm-1 to 282 cm-1 for Tc and from 274 cm-1 to 258 cm-1 for Re.
It has been recognized22 that the interrelationship of an M–M multiple bond with axial ligands (16.1) is quite comparable to that of multiple M–O and M–N bonds to trans ligands in octahedral complexes (16.2) and that the reasons, whatever they may be in detail, are presumably similar.
M>M–L |
X=M–L |
16.1 |
16.2 |
The W2(O2CR)4R'2 compounds23,24 constitute a special case. Here we find that in going from W2(O2CR)4 to W2(O2CR)4R'2 the W–W bond is not significantly lengthened, while the axial W–C bonds are fairly strong (c. 2.19 Å in length). Detailed calculations by the X_-SW method, with relativistic corrections25 have been carried out and provide a satisfactory explanation for this unusual behavior. They show that it arises because of properties peculiar to the tungsten atoms (with some possibility of molybdenum behaving similarly) and is not to be expected in general. The W– C axial bonds do not form in direct competition with the W–W μ-bond (as would normally be expected) because considerable 6s (and even some 6p) character is introduced. The molecular orbital mainly responsible for W–W μ bonding in W2(O2CR)4 is scarcely affected by the axial ligation in this case.
16.1.4 Comparison of second and third transition series homologs
There are several aspects to the comparison of second and third transition series homologs:
1.the relative stabilities of stoichiometrically analogous compounds (e.g., Mo and W, Tc and Re, Ru and Os, to mention the most common pairs),
2.the structural differences between pairs of homologous species when both can be isolated and characterized,
3.electronic structure differences.
There are some very striking differences in homologous pairs of compounds, while in many cases the differences are negligible. In comparing triply-bonded dimolybdenum and ditungsten compounds, there are few major stability differences. All types of M2X6 compounds are known for both elements, although a few specific differences do exist. For example, the metathesis reactions of W2(OCMe3)6 with acetylenes do not occur for any Mo2(OR)6 compound. There are more marked differences between the Mo-Mo and W–W quadruple bonds, however, consistently in the direction of the W24+ species being less stable, more easily oxidized, and more reactive generally. There are, however, many homologous pairs that exhibit considerable similarity, viz. the M2(mhp)4 and M2Cl4(PR3)4 compounds, and MoW4+ compounds are numerous and stable. Notably, no MoWX6 species has been made.
Greater susceptibility of W24+ compounds to attack by acids or other oxidizing agents is probably due to their having weaker β bonds, leaving the β electrons more open to attack. The W–W quadruple bonds are typically 0.1 Å longer than corresponding Mo–Mo bonds, and for the inherently small β overlap this might make a crucial difference. For the triple bonds, the W–W distances are also consistently greater, by 0.08-0.10 Å, but the μ and / electrons are so

714Multiple Bonds Between Metal Atoms Chapter 16
much more strongly held in both the Mo2 and W2 species that this may have little influence on their stabilities.
A few observations concerning differences in other groups are the following:
1.The reduction of [Tc2Cl8]2- to [Tc2Cl8]3- is easy, while conversion of [Re2Cl8]2- to [Re2Cl8]3- can be accomplished only below room temperature.
2.While Mo2(O2CCH3)4 is easily obtained by direct reaction of Mo(CO)6 with CH3CO2H, analogous chemistry does not occur for tungsten.
3.While over 1500 Rh24+ compounds are known and are generally easy to make, Ir24+ compounds of the M2X8 or paddlewheel of type are few. Whether thermodynamic instability or synthetic difficulties are responsible is an open question.
4.There are numerous singly-bonded Pt26+ compounds but only one Pd26+ complex, Pd2(hpp)4Cl2. Similarly, species with formal Pt25+ cores have been more extensively studied than those of the Pd analogs.
One generalization that clearly emerges from all of the above facts is that for the dinuclear species as well as for the conventional mononuclear complexes, higher oxidation states are favored for the third transition series as compared to the second. Among the reasons for this is the much greater magnitude of relativistic effects on ionization energies and spin-orbit coupling in the third series atoms.26
With particular respect to the formation of short M–M multiple bonds, it is of major importance that the third row elements, which follow the lanthanides, have very dense and relatively incompressible cores inside their valence-shell regions. The main consequence of the difference in core densities, which is displayed in Fig. 16.4 for molybdenum and tungsten, is that M–M bond lengths are affected strongly, but metal-ligand bond lengths are not. The M–L internuclear distances are much the same for Mo–L and W–L bonds in the M2 species, just as they are in mononuclear complexes, because the small ligand atom cores do not encounter the metal atom cores appreciably in either case. Thus, the valence-shell radii (Pauling’s R1 values) are practically the same. However, the core-core repulsions are significantly greater for W–W bonds than for Mo–Mo bonds, because for the former both of the bonded atoms have much denser cores than for the latter. This leads to the general result that for a given bond order (3 or 4) and the same or similar ligand sets, the W–W bond is 0.09-0.12 Å longer than the Mo–Mo bond.
Fig. 16.4. A comparison of the single-bond radii (Pauling R1 values) and core densities for molybdenum and tungsten atoms.

Physical, Spectroscopic and Theoretical Results 715
Cotton
In quadruply bonded heteronuclear Mo–W molecules, changes in the metal-metal distances are in qualitative accord with these considerations. Some pertinent results27,28 are:
Mo2(mhp)4 |
MoW(mhp)4 |
W2(mhp)4 |
2.065(1) Å |
2.091(1) Å |
2.161(1) Å |
|
¨ = 0.026(2) Å |
¨ = 0.070(2) Å |
Mo2(O2CCH3)4 |
MoW(O2CCH3)4 |
W2(O2CCF3)4 |
2.091(1) Å |
2.080(1) Å |
2.209(2) Å |
|
¨ = -0.011(2) Å |
¨ = 0.129(3) Å |
It is seen that the differences between Mo–Mo and Mo–W bond lengths are small and may be of either sign, whereas the change from Mo–W to W–W is consistently larger and positive.
16.1.5 Disorder in crystals
In addition to the kinds of crystallographic disorders that may occur in compounds of any kind, the tetragonal shapes of M2n+ molecules favor a characteristic type of orientational disorder in which the M2n+ unit within a cube-like set of ligands can display more than one orientation. This was first observed for the [Mo2Cl8]4- ion and is now known to occur very generally in M2X8n- ions and in some of their substitution products.
[M2X8]n- ions.
There is usually one primary orientation and one secondary one, but in a few cases there are two secondary ones. In two cases all three orientations are equally represented. Table 16.1 lists the pertinent compounds in which disorder has been reported.
Table 16.1. Orientational disorder in compounds of M2X8n- ions
Compound |
Occupancy (%) of orientations |
ref. |
||
(Bun4N)2Re2Cl8 |
74 |
26 |
|
29 |
(PHPr3)2Re2Cl8 |
76 |
24 |
|
30 |
(PMePh3)2Re2Cl8 |
61 |
39 |
|
30 |
(Et4N)2Re2Cl8 |
67 |
17 |
16 |
31 |
[ReCl2(depe)2]2Re2Cl8 |
74 |
26 |
|
32 |
(Bun4N)2Re2Br8 |
62 |
38 |
|
33 |
(PMePh3)2Re2Br8 |
82 |
18 |
|
30 |
(PPh4)2Re2Br8 |
95 |
5 |
|
34 |
(DMAA2H)2Re2Br8a |
57 |
36 |
7 |
35 |
(Bun4N)2Re2I8 |
33 |
33 |
33 |
36 |
(Bun4N)2Tc2Cl8 |
69 |
31 |
|
37 |
K4Mo2Cl8·2H2O |
90 |
10 |
|
38 |
(1,3-C3H6N2H6)2Mo2Cl8·4H2O |
53 |
47 |
|
38 |
(PMePh3)2Os2Cl8 |
63 |
37 |
|
39 |
[(C5Me5)2OsH]2Os2Br8 |
33 |
33 |
33 |
40 |
a DMAA2H = [(CH3CON(CH3)2)2H]
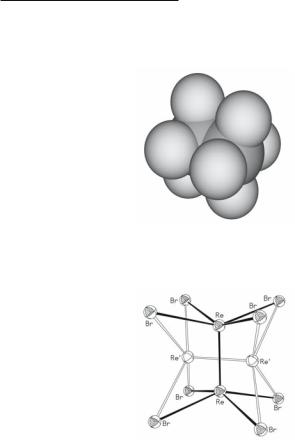
At the root of this type of disorder in [M2X8]n- compounds is the fact that these square parallelepipids are practically cubes, as shown in Fig. 16.5. They are only about 0.05 Å (c. 2%) higher than wide. Since vibrational amplitudes are greater than this, as sensed by its surround-
716Multiple Bonds Between Metal Atoms Chapter 16
ings in a crystal, an [M2X8]n- ion fits nearly as well in one direction as in either of the others. Disorder in the Re2Br82− ion,30 as shown in Fig. 16.6, is a typical example.
Fig. 16.5. A space-filling drawing of the Mo2Cl84- ion showing its practically cubic proportions.
Fig. 16.6. The 82:18 disorder in the Re2Br82- ion in (PMePh3)2[Re2Br8]
The positive charge on the M2n+ unit within the cube of X- ion is not uniformly distributed at all three opposite pairs of faces, and in general this leads to a preferred orientation of the M2X8n- ion. In about 70% of the reported structures there was no detectable secondary orientation.38,41 However, no quantitative correlation between the nature or distribution of the surrounding positive charges and orientational disorder of the M2X8n- ion has been established.38
The question of whether the extent of disorder (when it is not governed by crystallographic symmetry) is reproducible has been examined in a study of (Bun4N)2Re2Cl8.42 It was found that the percentages (74 and 26) are reproducible and are reliable to ± 0.5%.
Disordered crystal structures have also been found for some mixed-ligand species, M2XnL8-n. The largest class of mixed-ligand compounds are the neutral M2X4L4 molecules, where M = Mo, W or Re, X = a halide, pseudohalide or alkoxide ion, and L is usually a phosphine, although in a few cases L is an amine, alcohol, or nitrile. Only about 20% of the structures reported show disorder, and most (if not all) that do are listed in Table 16.2. All these molecules have the 1,3,6,8 distribution of ligands. Note that in three cases the disorder is complete (i.e., one third in each direction). Moreover, all of the disordered structures occur for molecules with L = PR3. In these cases the three identical R groups permit a certain “sloppiness” in their own orientations although generally the X and PR3 ligands each have their distinct locations. For the three M2Cl4(PEt3)4 molecules, the Cl and PEt3 ligands are also randomly disordered over the eight ligand sites.