

Metal-Catalysed Reactions of Hydrocarbons / 13-Reactions of the Lower Alkanes with Hydrogen
.pdf13
REACTIONS OF THE LOWER
ALKANES WITH HYDROGEN
PREFACE
We now approach the final chapter in which reactions achieving drastic restructuring of reactant hydrocarbons are to be considered. Some of these are of immense practical importance, while others, such as hydrogenolysis, are usually regarded as a nuisance, but these have attracted great interest from academic scientists and those in industry with an interest in fundamental studies. Hydrogenolysis is a difficult reaction in more senses than one: it requires somewhat high temperatures, even the most active metals performing only above about 373 K; it is ‘demanding’ in the sense of being regarded as structure-sensitive; it is (like most hydrocarbon reactions) bedevilled by ‘carbon’ formation; and its mathematical modelling has taxed the ingenuity of several generations of scientists.
We have met the breaking of C C bonds by hydrogen already in Chapter 11, but the molecules considered there (cyclopropane and cyclobutane) had some degree of alkene-like character and reacted easily (especially the former). In this chapter we shall be involved with linear and branched alkanes having two, three or four carbon atoms. C C bond fission is the principal process, but with the butanes skeletal isomerisation is also possible, and dehydrogenation sometimes happens at the same time. Reactions of acyclic and cyclic alkanes having five or more carbon atoms feature in the following chapter, where isomerisation and dehydrocyclisation are the important reactions. Some limited overlap between this chapters and the next is unavoidable.
We start with ‘A short philosophical digression’ that can be skipped if your interest does not run in that direction.
525

526 |
CHAPTER 13 |
13.1. INTRODUCTION
13.1.1. A Short Philosophical Digression
Physical scientists are not much given to philosophy. They tend to be down-to- earth people, prepared to believe what their senses tell them, and not very concerned about the wider philosophical implications. This reluctance is very clearly seen in the study of heterogeneous catalysis, where the construction of models and the drawing of inferences are exceptionally difficult but vitally important; the great variation in the vigour with which these actions are pursued reflects the uncertainty that scientists feel on departing from tangible observations.
The difficulties attending the extraction of an acceptable conceptual model for a reaction mechanism from experimental findings has been a recurrent theme of the last six chapters. A suggestion as to what needed to be defined before we could say we understood a reaction mechanism was made in Section 5.3; but this was only a start. Since in this chapter and the next the problems of resolving the nature of adsorbed intermediate and their modes of interaction becomes acute, it may be helpful to preface our discussion with a short reflection on exactly what it is we are trying to do, and how far our efforts are likely to meet with success.
The amount of information needed to start a discussion of mechanism is quite small: speculation can then proceed untrammelled by a superfluity of experimental facts. This was often the case with early works on many systems, but not surprisingly as further observations were made the model had to be refined and extended: and it became more complicated. More assumptions and suppositions had to be introduced, and more uncertainty entered the conclusions.
An example of a complicating refinement lies in the choice of terminology and symbolism used to represent our ideas. One started with statements employing letters as chemicals symbols, e.g. H for the hydrogen atom; but this gives no idea of its size relative to that of other atoms, and it is easy to come to think that a hydrogen atom really is a capital aitch. This kind of symbolism is quite unsuited for describing what goes on at metal surfaces or the structure of imagined adsorbed intermediates. Scale drawings on paper or a visual display unit are essential for accurately representing adsorbed species, but even this is not quite the end, because there is then the need to show their dynamic interaction both in geometric and energetic dimensions. Dynamic computer simulation is the only way to manage the first of these. There is truly no end to the number of layers of the mechanistic onion!
If correct logic is applied, we must suppose that every mechanistic statement is valid within the confines of the theoretical concepts and symbolism available at the time, and the accessible experimental conditions and techniques. The virtuous circle of experiment → model → experiment, which is supposed to characterise the scientific method, seems to be followed less often than it might, and

REACTIONS OF THE LOWER ALKANES WITH HYDROGEN |
527 |
one rarely reads about predictions that a particular model suggests. A common failure is the disregard of relevant results from analogous systems, and the NIH (not invented here) factor is often perceived in ‘Discussion’ sections of papers. Under-interpretation is more usual than over-interpretation. Popper’s dictum has been mentioned several times: it is necessary to appreciate that one’s mechanistic statement is not the last word, but should be used as a springboard to design the experiments to test it. Self-doubt is not a sin, and it is better to be critical oneself of one’s conclusions than to await the criticisms of others. Incidentally, Popper’s admirable philosophical position was anticipated by Alfred, Lord Tennyson:
For nothing worthy proving can be proven
Nor yet disproven; wherefore be thou wise:
Cleave ever to the sunnier side of doubt.
Scientists and poets throughout the ages have been aware of the conflict between the mass of available information and the understanding of its significance. J.W. von Goethe summed it up succinctly: We know accurately only when we know little; with knowledge, doubt increases. This may be paraphrased as: The more we know, the more we know we don’t know. Somewhat more optimistically, Francis Bacon, Lord Verulam, believed that If a man begin with certainties, he shall end in doubts; but if he will be content to begin with doubts, he shall end in certainties.
13.1.2. Alkane Hydrogenolysis: General Characteristics
In the context of alkanes, hydrogenolysis is the breaking of C C bonds by the action of hydrogen, leading to alkanes of lower molar mass. It is not a reaction that is deliberately practised on a large scale, but it is a parasitic reaction that occurs in parallel with other useful reactions of alkanes to be considered in the next chapter. In order to learn how to avoid or minimise it, it becomes necessary to find out as much as possible about it, and this is most easily done with molecules containing only two to four carbon atoms. Alkanes can also be ‘cracked’ by an acid-catalysed reaction on solid acids or acidic supports, but in this chapter we are solely concerned with reactions that proceed on purely metallic sites: the cooperation of metallic and acidic sites in petroleum reforming will be briefly considered in Chapter 14.
The reaction has also proved interesting because of its reputation for structuresensitivity. There have been numerous studies of the hydrogenolysis of alkanes (especially ethane, which has the merit of giving only a single product), examining their response to variations in particle size, and ensemble size in bimetallic systems, to face-sensitivity with single-crystal surfaces and to the Strong Metal-Support Interaction. The reactions are exothermic, but need temperatures that generally exceed 373 K because activation energies are high. It is generally believed that a multi-atom site is necessary in order to accommodate a species in which two or

528 |
CHAPTER 13 |
more C M bonds from different carbon atoms are formed, as these are needed to produce the strain in the C C bond that causes it to break. This of course demands the breaking of several C H bonds, and sites to receive the hydrogen atoms thus released; this apparently accounts for the very large negative orders in hydrogen that are often observed. The complex history of the attempts to create mathematical models to describe the kinetics of this class of reaction will be surveyed in Section 13.2.
13.1.3. Problems in Studying Reaction Kinetics
The conditions under which hydrogenolysis occurs readily are generally speaking those that favour ‘carbon’ deposition.1 This can be limited by the use of high hydrogen: alkane ratios, and many studies have been conducted using ratios of 10 or more. However, every silvery lining must have its cloud, and the disadvantage of doing this is that the negative order in hydrogen means that rates are lower than they might have been if a lower ratio had been used. There is a paradox here that cannot be avoided. The reactive form of the alkane is partially dehydrogenated, and moving to high hydrogen/alkane ratios makes its formation less likely, but it is also the form that at low ratios leads to the formation of ‘carbon’ and hence to deactivation. You can’t win. There are therefore major problems in obtaining meaningful kinetic results, and it is worth emphasising that for purposes of kinetic modelling it is very important to have results over as wide a range of reactant ratios as possible, and particularly to establish the co-ordinates of the rate maximum. It therefore becomes necessary to seek ways of overcoming this problem.
The injection of hydrocarbon pulses into a hydrogen stream has been widely practised, and reveals the activity of metal catalysts and the reactivity of alkanes under standard conditions,2 but does not allow determination of reaction orders. One procedure that has proved suitable for acquiring good kinetic results is the short reaction period (SRP) method,3,4 in which an alkane-hydrogen mixture of known composition is passed over the catalyst for 1 min and sampled; hydrogen is then passed for 19 min to cleanse the surface, and then another SRP started. This minimises or even eliminates the loss of activity experienced when continuous flow of reactants is used; in the n-butane-hydrogen reaction over PtRe/Al2O3 at 582 K, it even leads to some increase in activity.3 Variations on this standard procedure have been introduced when it has been desired to study the effects of ‘carbon’ laydown. When deactivation does occur, further improvement in the quality of results may be obtained by having frequent recourse to a standard reactant concentration, and adjusting the rate found with a different reactant ratio by interpolation, remembering that this rate is fixed by the previous standard value. Only with zeolite-supported platinum did these procedures not work, because hydrocarbon pulses were totally retained.5 For further details, the cited references should be consulted.

REACTIONS OF THE LOWER ALKANES WITH HYDROGEN |
529 |
Figure 13.1. Hydrogenolysis of n-butane on 1.5% Re/Al2 O3 : Arrhenius plots for two successive thermal cycles.6
The distinction between lowered activity because of (i) ‘carbon’ formation and (ii) inappropriate concentrations of reactive species is established by the subsequent use of a standard reactant mixture, but sometimes extent of the problem depends unexpectedly on temperature and on the direction of temperature change. Rhenium is a very active metal for hydrogenolysis, and the variation in rate of the n-butane-hydrogen reaction as temperature was raised and then lowered in a stepwise manner is shown in Figure 13.1.6 Initially some ‘carbon’ formation occurs, and this leads to a small rate maximum, but then as temperature is raised further it is gradually removed and the reaction continues with a higher activation energy. On lowering the temperature the surface remains in a clean state and a smoothly decreasing rate is observed. This effect may be described as hysteresis caused by ‘carbon’ formation. In a second thermal cycle, ‘carbon’ formation starts sooner, but the rates found with descending temperature exactly reproduce those in the first cycle.
The tendency to deactivate by ‘carbon’ formation runs parallel to activity in hydrogenolysis because the same or similar intermediate species are involved. Thus platinum, with which most work has been done and which has been chosen as best for other desirable reactions (dehydrogenation (see Chapter 12), isomerisation etc.) because of its low activity for hydrogenolysis, is much easier to work with than the base metals. On this metal, and probably generally, ease of forming the multiply-bonded intermediates increases with the number of carbon atoms, since as chain-length increases, so does the chance of C H bond fission, and not all the dehydrogenated species are those needed for hydrogenolysis.7,8 So with n-butane, a 1,1,2-triadsorbed species may lead to the breaking of the terminal C C bond, whereas a 1,2,3-triadsorbed species may end up as ‘carbon’. Consequentially little deactivation is experienced when ethane reacts over platinum, but problems mount as the chain-length grows.

530 |
CHAPTER 13 |
13.1.4. Ways of Expressing Product Composition
Ways in which rates of reaction may be expressed have been considered in Section 5.2.3. In the present context they are usually given in specific or areal terms, or as turnover frequencies, i.e. rate per (presumed) active centre. Where standard catalysts such as EUROPT-1 have been used, the rate per unit mass of catalyst or metal is sufficient, because this can readily be translated into areal units if comparison with other catalysts is desired.
There is much greater variation in the means used to express product selectivities. This is a very important aspect of research in this area, because the nature and amounts of the products formed is determined by the type and reactivities of the adsorbed intermediates, and hence reveals intimate features of the reaction mechanism. The larger and more complex the structure of the reactant alkane, the greater is the need for clarity. Giving the fraction of each product as a percentage of the total9 is not very useful, neither is the use of ratios of pairs of products,10 because the overall picture is obscured. The Budapest group have employed a fragmentation factor (ζ )11 defined as
ζ = Ci / (i/n)Ci |
(13.1) |
where Ci is the amount of product containing i carbon atoms formed from a reactant having n carbon atoms. Thus ζ is two for a single C C bond splitting and n where complete breakdown to methane occurs. This parameter must apply only to initial product yields, as it will increase with reaction progress as first-formed products react further. The products of complex alkanes containing different types of C C bond have been characterised by a reactivity factor (ω) that defines the chance of a particular bond breaking relative to its statistical chance.12 By far the most useful procedure for the lower alkanes is simply to define the selectivity to the molecule having j carbon atoms (S j ) as
|
S j = C j / A |
(13.2) |
where C j is the molar fraction of that product and |
A the moles of reactant |
|
converted.3,13–16 This leads to the relations17 . . . . |
|
|
For propane: |
S1 + 2S2 = 3 |
(13.3) |
For n-butane: |
S1 + 2S2 + 3S3 = 4 |
(13.4) |
Defined thus, the selectivities can be directly fed into equations derived from a general mechanistic scheme, yielding more fundamental parameters, as will be disclosed shortly. When isomerisation of n-butane is observed, the fraction reacting

REACTIONS OF THE LOWER ALKANES WITH HYDROGEN |
531 |
in this way is denoted by Si , and the other selectivities are based on the remaining fraction.
13.2.HYDROGENOLYSIS OF THE LOWER ALKANES ON SINGLE METAL CATALYSTS: RATES, KINETICS, AND MECHANISMS
13.2.1. The Beginning
The orderly presentation of the very extensive literature falling under this heading presents considerable difficulties. According to Max Planck, The chief problem in every science is that of endeavouring to arrange and collate the numerous individual observations and details which present themselves, in order that they may become part of one comprehensive picture. Let us see how this problem might be addressed.
The information available is of two types. (1) First is the dependence of rate on the kind of alkane, on the nature of the catalyst, on reactant concentrations and on temperature. (2) Second is the dependence of product selectivities or other descriptive factor18 (for molecules having three or more carbon atoms) on these variables. This separation is somewhat arbitrary, but the first type focuses on the rate-determining step, and leads us into a discussion of mechanism that lacks the refinements needed to understand the origin of selectivities and their variation with conditions. Some people have combined these two features by evaluating orders of reaction and activation energies for the formation of each individual product,19−22 and assume by implication that each reaction that can be formulated to give these products demands a different sort of site: but the numbers that emerge can only be used in a qualitative way, and the differences in site architecture cannot be defined. This approach is therefore somewhat sterile, and the alternative, which is to treat the rate of product removal and product selectivities separately, is to be preferred and is the one adopted here.
References to relevant reviews are collected in the Further Reading section at the end of the chapter.
13.2.2. Kinetic Parameters
It is convenient to start by considering the results obtained many years ago by John Sinfelt23 and his associates for the hydrogenolysis of ethane;24−29 they are models of clarity, and they provide a suitable framework for results on other systems. In terms of activity, metals divide themselves easily into three classes:
(i) the very active (ruthenium and osmium); (ii) the moderately active (the majority); and (iii) the least active (palladium and platinum). This distinction is clearly drawn when the Arrhenius parameters are depicted as a compensation plot
532 |
CHAPTER 13 |
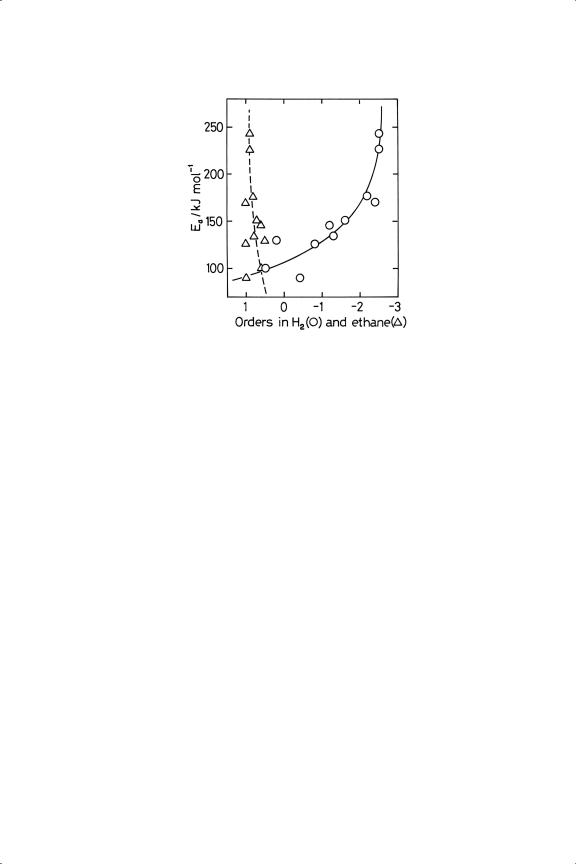
Figure 13.2. Hydrogenolysis of ethane: orders of reaction as function of activation energy for various silica-supported metals.24,25
(Figure 5.15);30 it is however necessary to draw the lines appropriately.25 It was suggested earlier (Section 5.6) that metals sharing a single compensation line might operate by a common mechanism, or at least have some mechanistic features linking them: by definition there must be some temperature (the isokinetic temperature) at which they all show the same rate, and for the majority of metals this should be at about 383 K. However at any other temperature their rates will vary as determined by the Arrhenius parameters between the activation energies and the orders of reaction. The orders in hydrogen, which were in most cases negative, tended to become more positive as the temperature of measurement was necessarily increased due to lessening activity, but palladium and platinum were exceptions. There is a good correlation between activation energy and hydrogen order, and a less good correlation with ethane order (Figure 13.2):30 the least active metals showed (not surprisingly) highest activation energies, but also most negative orders in hydrogen. This frequently observed inhibition by hydrogen has always been taken to imply that the hydrocarbon has to lose a certain number of hydrogen atoms to be activated and thus to need a certain number of free surface sites. Strong hydrogen chemisorption would thus run in parallel with weak alkane chemisorption (Figure 13.2). The results suggested that the adsorption of hydrogen on palladium and platinum must be exceptionally strong, a possibility that was mooted for platinum in the previous chapter in the context of dehydrogenation. This qualitative picture will be quantified in a later section.
Similar if less clear categorisation of metals according to their activities is possible using results obtained with ethane hydrogenolysis by metal blacks,31 and with

REACTIONS OF THE LOWER ALKANES WITH HYDROGEN |
533 |
n-pentane hydrogenolysis on silica-supported metals.32 However in both these cases the databases are smaller.
Very extensive results are available for the hydrogenolysis of alkanes on many of the metals of Groups 8 to 10. Only iron, cobalt and osmium have been little studied, the first because of its great propensity to destructive adsorption of alkanes. Activation energies have frequently been reported (although their precision is quite rarely given), but reliable pre-exponential factors (i.e. based on specific rates or TOFs) are less common, especially in early work where the metal dispersion was often not determined. Their accurate estimation also demands a precise knowledge of the metal content of supported metals, and assuming the truth of the ‘nominal’ content is a frequent source of error. Much less often are orders of reaction given, and then usually at only a single temperature; some of these orders are quoted in Table 13.2. More detailed studies employing wider ranges of conditions will be the subject of a later section. Consideration of the Arrhenius parameters, with all their faults, does however convey some useful messages.
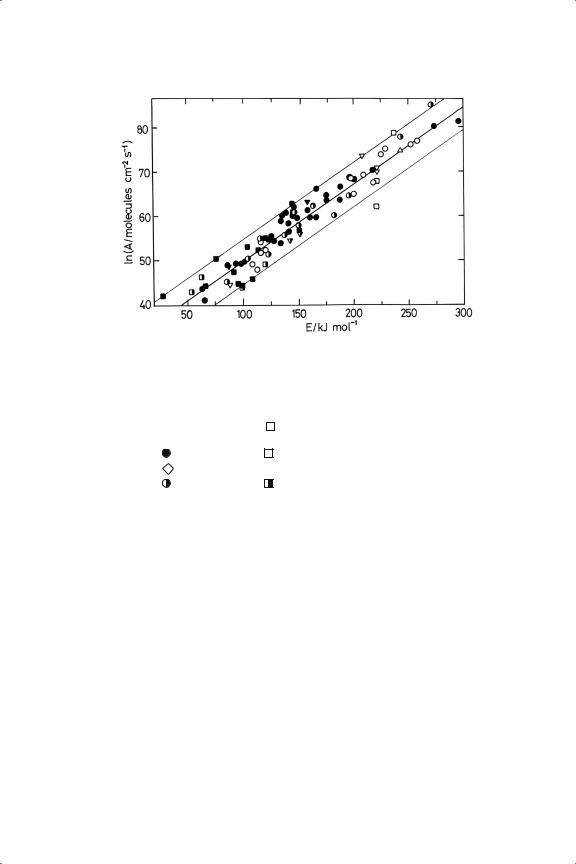
These are most economically summarised as compensation plots. We may start by considering the many results available for the hydrogenolysis of the lower alkanes on various platinum (and palladium) catalysts:30,33−35 the Arrhenius parameters lie within a band about a line corresponding to an isokinetic temperature of about 590 K,33 the activation energies varying by almost a factor of six ( 50 to300 kJ mol−1, Figure 13.3). Some of the values shown in this figure were included in the original compilation,33 but most are new or newly discovered. The collections of results made by Gabor Somorjai and his colleagues34,35 have been a great help in this respect. At first sight the data points lie randomly within the band, irrespective of alkane structure, type of platinum catalyst or experimental conditions: those in Figure 13.3 cover ethane, propane and the butanes, and there are a few points relating to C5 alkanes. The sources of the information are listed in the figure legends. It is likely that the vertical width of the band is substantially due to errors in estimating metal area, although an error of ±20% on TOF converts to only ±0.2 on ln A, i.e. about the size of the points on the figures. This source of error is removed when the same catalyst (EUROPT-1, 6.3% Pt/SiO2) is used to obtain results for the total reaction of C3 to C6 alkanes and neopentane, points for which lie almost exactly on a single line, while those for the individual hydrogenolyses and isomerisations lie about a separate lower line (Figure 13.4).33 Points for ethane hydrogenolysis (not shown) are on a yet lower line.33 A further reason for the variability in Figure 13.3 (but absent from the result in Figure 13.4) might be the very different reactant pressures used; hydrocarbon pressures have varied by at least 102, from 10−3 atm in UHV work to 0.15 atm in some flow-systems, and it remains to be seen whether the effect this would have on rate is accompanied by changes in the Arrhenius parameters.
We may therefore conclude that on platinum there are no radical differences in mechanism as chain length is increased, and that hydrogenolysis and skeletal
534 |
CHAPTER 13 |
Figure 13.3. Compensation plot of Arrhenius parameters for alkane hydrogenolysis (mainly C2 to C4 ) on various platinum and palladium catalysts. 1 = Pt; 2 = Pd.
|
|
Symbols used in Figures 13.3 and 13.4–13.8 |
||||
Metal |
Supported |
Unsupported |
Alkane/Process |
|||
—————————————————————————————— |
||||||
1 |
|
|
C2 H6 |
|||
2 |
|
etc. |
|
|||
1 |
|
|
|
|
>C2 |
/total or hydrogenolysis only |
|
|
|||||
3 |
|
etc. |
|
|||
1 |
|
|
|
|
>C3 |
/isomerisation only |
The units of A are molecules cm−2 s−1 . Where rates or values of A are given in units of time−1 , i.e., where a number of ‘sites’ or surface atoms has been estimated, they have been changed by assuming 1015 ‘sites’ cm−2 : the mean value based on Pt(111) and Pt(100) is close to 1.5 × 1015 , but the same round figure is taken as a mean for all metals of Groups 8 to 11.
isomerisation are processes that are closely linked. Ethane does however distinguish itself from the higher alkanes.
Closer examination of the location of points within Figure 13.3 and the tables of results on which it is based lead to the following further conclusions. (i) While the points for single crystals lie well within the band, those for metal ‘blacks’ are towards the bottom or even below it, perhaps because their effective areas have been over-estimated. (ii) Activation energies for the alkanes usually decrease with increasing chain length, from about 200–250 kJ mol−1 for ethane to a lower limit reached at about C5 or C6 (Table 13.1); values for isobutane are generally higher than those for n-butane, and those for isomerisation are often (but not always) higher than those for hydrogenolysis (see Figure 13.4). As might be expected from Figure 5.15, the few additional points for palladium lie within the platinum band. Note that in this discussion, as in much that has gone before, ‘activation energy’