

Metal-Catalysed Reactions of Hydrocarbons / 08-Hydrogenation of Alkenes and Related Processes
.pdf8
HYDROGENATION OF ALKADIENES AND POLY-ENES
PREFACE
In this and the next chapter the principal theme will be the selective partial reduction of an unsaturated hydrocarbon to a desired intermediate product, usually an alkene, and, connected with this, its selective reduction in the presence of an excess of the alkene; this represents an important industrial problem, to the solution of which much academic effort has been devoted. The formal framework for selectivity in heterogeneous catalysis has been presented in Chapter 5; our task is now to see how those principles apply, first, to the hydrogenation of dienes, and then in the next chapter to the hydrogenation of alkynes. In the case of dienes, different modes of addition of hydrogen atoms result in different products, and these modes are sensitive to the metal used and to other experimental variables. The task of defining the mechanisms by which products are formed, and of identifying probable intermediates has been assisted by the application of isotopic labelling by the use of deuterium, and a number of studies have sought to illuminate the role of the surface composition in determining the origin of selectivity by using bimetallic and selectively poisoned catalysts. From this work there has been obtained a quite high state of understanding of the ways in which the hydrogenation of dienes proceeds.
8.1. INTRODUCTION
8.1.1. Types of Unsaturation
The principal subject of this chapter is the hydrogenation of molecules containing two carbon-carbon double bonds. These may exist in a variety of situations: thus in a linear chain of carbon atoms they may be adjacent as in propadiene
357

358 CHAPTER 8
TABLE 8.1. Heats of Hydrogenation of Multiply-
Unsaturated Hydrocarbons (kJ mol−1 )
Alkynes |
− |
HH |
Ethyne→ethene |
174 |
|
Propyne→propene |
163 |
|
1-Butyne→1-butene |
165 |
|
2-Butyne→Z-2-butene |
153 |
|
2-Butyne→E-2-butene |
157 |
|
|
|
|
Alkadienes |
− |
HH |
Propadiene→Propene |
172 |
|
1,2-Butadiene→1-butene |
162 |
|
1,2-Butadiene→Z-2-butene |
169 |
|
1,3-Butadiene→1-butene |
110 |
|
|
|
|
Cycloalkenes and cycloalkanes |
− |
HH |
Cyclopropene→cyclopropane |
224 |
|
Cyclopropane→propane |
157 |
|
Cyclobutene→cyclobutane |
129 |
|
Cyclobutane→ n-butane |
153 |
|
See Physical Properties of Hydrocarbons (R.W. Gallant and C.L. Yaws, eds.), 2nd . edn., Gulf Publ. Co.: Houston, Texas (1992) and ref. 25.
(allene), conjugated as in 1,3-butadiene or separated by one or more methylene groups as in 1,4-pentadiene. The closer they are together, the more they interact;1 thus the heat of hydrogenation of allene to propene is greater than that for propyne to propene (Table 8.1), while that of butadiene to 1-butene is typical of the values for alkenes (see Table 7.1). So when one or more methylene groups separates the double bonds, we may expect them to act independently in chemical terms, although it is of course possible for both to be chemisorbed at the same time, and on metals that are effective for double bond migration (such as palladium) they may move into the stabler conjugated state, so that the major products will be the same as those given by the 1,3-diene. Pairs of double bonds of any of these types may exist in cyclic molecules (Sections 8.2.2, 8.2.3 and 8.4.3). In terpene and steroid chemistry, cyclic dienes are frequently met,2,3 and their hydrogenation follows complex pathways, especially when isomerisation is possible; we shall not enter far into this minefield.
Triplets or quartets of double bonds can also be found in linear hydrocarbons and in larger ring systems (e.g. C10 or C12); the principles outlined above also apply to them (Section 8.4.3). Compounds in which three or more double bonds exist adjacently, either in linear or cyclic systems, are referred to as cumulenes; they show some interesting features when being hydrogenated.3,4

HYDROGENATION OF ALKADIENES AND POLY-ENES |
359 |
8.1.2.Practical Applications of Selective Hydrogenation: Outline of Mechanisms5–7
Various dienes and alkynes are significant components of the products formed by the steam-cracking of naphtha designing to give streams of the lower alkenes (C2 to C4) for further petrochemical manipulation, such as selective oxidation or polymerisation. The main type of alkene made, and the nature and concentrations of the polyunsaturates, depends on the severity of the steam-cracking. The presence of these polyunsaturates is highly undesirable, as they interfere with subsequent processing of the alkenes by reason of either their ready oxidation or oligomerisation (in the case of dienes) or their toxicity towards catalysts (in the case of alkynes). It is therefore essential to treat these streams to remove them to very low limits (generally to ppm), ideally by reducing them to useful alkenes, but without hydrogenating any of the alkene, which is present in great excess, or causing its isomerisation. This is a taxing practical matter of very great industrial importance,7,8 which has spawned an enormous patent literature, and has attracted the attention of a number of academic laboratories, motivated by the desire to understand the basics of selective hydrogenation and to discover ever more effective catalysts. In laboratory work, 1,3-butadiene has been undoubtedly the model compound of choice, and there is very extensive literature on its hydrogenation;9−14 the term ‘butadiene’ therefore is taken to mean the 1,3-isomer so often will the word be used. Corresponding studies on the selective hydrogenation of alkynes will be considered in Chapter 9.
The formal representation of selective processes was introduced in Chapter 5 (Section 5.2.6). In the present context the successful selective hydrogenation of a diene to an alkene demands that (i) the alkene formed from the diene should vacate the surface rather than awaiting reduction to the alkane, and (ii) the stronger chemisorption of the diene should totally exclude the alkene from the surface, so that it emerges unscathed. When a diene is used by itself, the reaction will cease entirely when it is completely hydrogenated by one mol of hydrogen per mol of diene if the catalyst is perfectly selective. These are counsels of perfection, but catalysts based on palladium have a long history of successful use in this process. The extent of their success is measured by the residual content of the diene and the loss or gain in the concentration of the alkene,15 as well as by conventional criteria such as longevity. In the broader context, i.e. away from alkene manufacture, selectivity can take two other forms:2,3 (i) chemoselectivity, where a C C bond is hydrogenated rather than some other potentially reducible function such as >C
C bond is hydrogenated rather than some other potentially reducible function such as >C O or -NO2, and (ii) regioselectivity, where the reduction of one C
O or -NO2, and (ii) regioselectivity, where the reduction of one C C bond is favoured over that of another in different surroundings: so for example a terminal bond is often more easily reduced than an internal one.
C bond is favoured over that of another in different surroundings: so for example a terminal bond is often more easily reduced than an internal one.
One of the major themes of basic research having a practical orientation has been the use of bimetallic catalysts for improved selectivity. Palladium is the

360 |
CHAPTER 8 |
predominant active metal that features in almost all industrial uses, and numerous additives have been tried, with some success. Organic molecules, particularly those containing nitrogen16 or sulfur,17 act as selective poisons, being less strongly adsorbed than dienes but more strongly held than alkenes.
One very major industrial application of selective hydrogenation is fat hardening.18−21 This subject will not be dealt with in detail, as the molecules being used are triglycerides, that is, esters of glycerol and unsaturated long-chain fatty acids (mainly having 18 carbon atoms); such a chain does however behave much like a hydrocarbon. The object of the game, put simply, is to reduce selectively two of the three C C bonds which occur in each chain of many animal and vegetable oils, without causing the remaining one to change from Z to E. This process is carried out under conditions of mass-transport limitation by supported nickel catalysts, although the use of palladium has been proposed.19,21 It alters materials that are easily autoxidisable to stable products that are edible (e.g. margarine) or have culinary use. High selectivity requires that the double bonds, not initially conjugated, rapidly become so, otherwise full hydrogenation would result; this determines the choice of catalytic metal. Copper also gives selective reduction, but is unsuitable because trace amounts of copper in the product destabilise it.
C bonds which occur in each chain of many animal and vegetable oils, without causing the remaining one to change from Z to E. This process is carried out under conditions of mass-transport limitation by supported nickel catalysts, although the use of palladium has been proposed.19,21 It alters materials that are easily autoxidisable to stable products that are edible (e.g. margarine) or have culinary use. High selectivity requires that the double bonds, not initially conjugated, rapidly become so, otherwise full hydrogenation would result; this determines the choice of catalytic metal. Copper also gives selective reduction, but is unsuitable because trace amounts of copper in the product destabilise it.
Our main task in this chapter is to try to understand how, when and why dienes and their relatives chemisorb to the exclusion of alkenes (the reasons are not always straightforward), and to explore the mechanisms that, additional to those revealed in the last Chapter, account for the formation of the observed products.10,14,22,23
8.2. HYDROGENATION OF 1, 2-ALKADIENES (ALLENES)
8.2.1. Hydrogenation of Propadiene
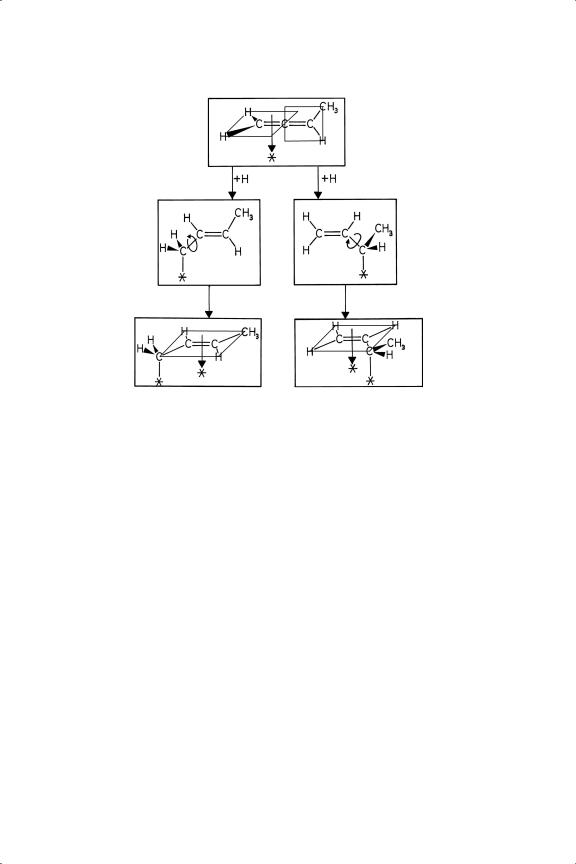
Little is known by direct experimentation of the manner in which propadiene is chemisorbed at metal surfaces.24 At low temperature (90 or 130 K) it is held on Cu(110) and on silver film as a molecular species in which the orthogonal π-systems are oriented so that one methylene group is normal to the surface and the other is parallel to it:25,26 on Ni(111) however it isomerises to di-σ-di-π-propyne27 (see structure 15 in Table 4.2). This arrangement of the two sets of π -orbitals at right-angles to each other means that compounds of the type R1R2C C
C CR3R4 have non-super imposable mirror images (Figure 8.1) and are therefore optically active:28 the C3 unit therefore behaves just as a single carbon atoms does. It would therefore appear that simultaneous chemisorption of both bonds as π-species at a flat surface is improbable, although strong bonding through one, and weak bonding through the other, is not impossible: but a tetra-σ species is unlikely to be seen except possibly at steps. Bonding through just one of the two double-bonds is therefore most likely: study of propadiene’s chemisorption on single crystal
CR3R4 have non-super imposable mirror images (Figure 8.1) and are therefore optically active:28 the C3 unit therefore behaves just as a single carbon atoms does. It would therefore appear that simultaneous chemisorption of both bonds as π-species at a flat surface is improbable, although strong bonding through one, and weak bonding through the other, is not impossible: but a tetra-σ species is unlikely to be seen except possibly at steps. Bonding through just one of the two double-bonds is therefore most likely: study of propadiene’s chemisorption on single crystal
HYDROGENATION OF ALKADIENES AND POLY-ENES |
361 |
Figure 8.1. Formation of πσ half-hydrogenated states in the hydrogenation of 1,2-butadiene.
surfaces, and of its coordination in complexes of the chlorides of Groups 9 and 10 metals, are lacunae that deserve to be filled.
The propadiene molecule is clearly quite highly strained, because the stabilisation that follows the opening of one of the two double bonds is very considerable. Its heat of hydrogenation to propene is greater than that for propyne (Table 8.1), and an early study29,30 showed that it is hydrogenated to propene on pumice-supported metals of Group 10 with high selectivity, especially on nickel and palladium: kinetic parameters are summarised in Table 8.2. In admixture with either ethyne or propyne, it was hydrogenated at similar rates, and neither propene nor cyclopropane interfered with its reduction.31 Kinetic parameters for the reaction over all the Group 8 to 10 metals as powders are available32−34 (Table 8.2): selectivities to propene were always high (>0.95), even for iridium. For nickel-copper bimetallic powders, the rate was maximal at 10–20% copper, but the activation energy was constant at 25 kJ mol−1 for 0–50% copper, then rising to 49 kJ mol−1 for pure copper.35 Temperature-programmed reaction on Pd(100) pre-covered with hydrogen has confirmed its selective reduction to propene,36 but on Pd/Al2O3 at 293 K the reaction was much less selective.37
During the reaction, part of the propadiene is converted into products of higher molar mass;30,32 these are linear and branched C6 products.37 Their formation has been explained30 by isomerisation of the half-hydrogenated state

362 CHAPTER 8
TABLE 8.2. Kinetic Parameters for the Hydrogenation of Propadiene on Pumice-Supported Metals r PH x PP y
Metal |
x |
y |
E /kJ mol−1 |
References |
Fe |
1.5 |
— |
39 |
34 |
Co |
1 |
— |
45 |
34 |
Ni |
1 |
0 |
54 |
29,30 |
|
1 |
— |
33 |
34 |
Ru |
1.4 |
— |
13 |
34 |
Rh |
0.8 |
— |
40 |
34 |
Pd |
1 |
>0 |
51 |
29,30 |
|
1 |
— |
54 |
34 |
Pd |
1.1 |
−0.4 |
29 |
37 |
Os |
0.9 |
— |
19 |
34 |
Ir |
1 |
— |
22 |
34 |
Pt |
1.1 |
<0 |
71 |
29,30 |
|
1 |
— |
73 |
34 |
Note: references 32 and 33 give somewhat different activation energies for the reaction on metal powders.Reaction with D2 on Pd/A12 O3.
(2-propenyl) gives a free-radical form that can attack a neighbouring adsorbed propadiene molecule to give a C6 species, but further chain-propagation does not occur.
Propadiene reacted with deuterium over Pd/Al2O3 at 293 K to give deuterated propenes and propanes, but neither exchanged propadiene nor hydrogen deuteride was observed;37 the concentration of adsorbed hydrogen and deuterium atoms was therefore low, and propadiene must have been strongly adsorbed. Burwell’s N profile method36−39 was applied to the distribution of deuteropropenes. This procedure is based on the assumption that there is a ‘pool’ of hydrogen and deuterium atoms, and that each position in the hydrocarbon equilibrates with this pool: from the observed distribution, the deuterium content of the pool a and the N profile (where Ni is the fraction of molecules in which i positions have equilibrated) can be deduced. In this case, a for propene formation was 83 ± 2%, this figure being confirmed by determining by NMR spectroscopy the ratio of the two isotopes on the central carbon atom. If the direct transfer of atoms between adsorbed species is possible, as discussed in Chapter 7, then the ratio a/(1-a) represents the integral over all contributing processes. The value of a for propane formation is smaller (77%), suggesting that different processes are involved in its formation.
8.2.2. Hydrogenation of Substituted 1, 2-Alkadienes
The reaction of 1,2-butadiene with hydrogen and with deuterium has been followed on palladium25,37,40 and nickel26.40 catalysts, with results that are summarised in Table 8.3. Those for the two palladium catalysts differ somewhat, e.g.

HYDROGENATION OF ALKADIENES AND POLY-ENES |
363 |
TABLE 8.3. Reaction of 1,2-Butadiene with Hydrogen and Deuterium: Product Composition and D Content of ‘Pool’
Catalyst |
1-Butene |
Z -2-Butene |
E -2-Butene |
D/% |
References |
Pd/A12 O3 |
40 |
53 |
7 |
94 |
25 |
Pd/A12 O3 |
40 |
60 |
0 |
81 |
26,40 |
Ni/SiO2 |
38 |
57 |
5 |
84 |
26,40 |
Ni/A12 O3 |
26 |
44 |
30 |
93 |
26,40 |
Ni powder gave similar results.
in respect of the yields of the d2-butenes and in the values of a; some effect of metal particle size or other characteristic of the catalyst was likely responsible. Although only a small fraction of the reactant was lost by dimerisation, in the case of the nickel catalysts26 no less than half disappeared in this way, and the different types exhibited two quite different behaviours. In each case there was little exchange in the reactant, but some isomerised giving mainly 1,3-butadiene-d0 as the result of an intramolecular hydrogen atom transfer. Nickel powder and Ni/SiO2 however gave 1-butene as the major product (Type A mode), while Ni/Al2O3 gave mainly E-2-butene (Type B mode, Table 8.3). A similar dichotomy has been seen in the reaction of 1,3-butadiene (Section 8.3.4), and the eccentric behaviour of Ni/Al2O3 is now known to be a consequence of its being contaminated with sulfur. It is believed that this encourages the conversion of π σ half-hydrogenated states into π-alkenylic species, and that this opens an additional channel through which E-2-butene can be formed. Of the various possible ways in which the reactant might chemisorb, the most probable is that shown in Figure 8.1. An alternative explanation of the effect of sulfur is that for some reason it encourages the insertion of the first hydrogen atom at the central unsaturated carbon atom, and that the σ -adsorbed butenyl species can then rotate freely as shown in Figure 8.1, adopting the preferred E-conformation before the remaining π-bond engages the surface and the σ C––M bond is broken.
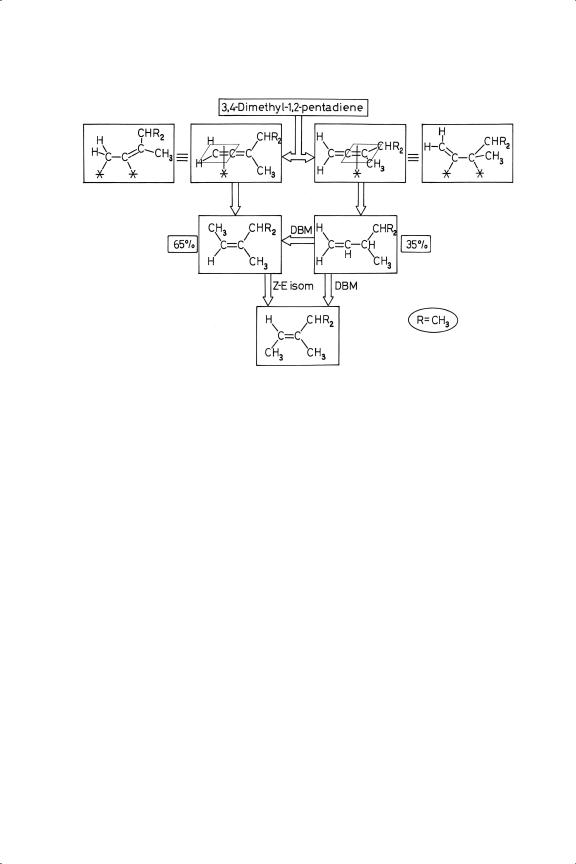
The importance of alkyl substituents about the pair of adjacent double bonds in determining preferred orientations in the chemisorbed state (or attitudes of presentation41) is well illustrated by the extensive work of Crombie and his colleagues, using 5% Pd/BaSO4 as catalyst. Their results can be illustrated by reference to 3,4-dimethyl-1,2-pentadiene(I), 4-methyl-2,3-hexadiene (II) and 3,4,4- trimethyl-1,2-pentadiene (III). Formation of the final alkane was usually slight until the diene had been wholly removed, although with III, which has an isobutyl substituent, it started earlier, probably because the reactant molecules could not pack so tightly, and the concentration of adsorbed hydrogen atoms was in consequence higher. Similarly, isomerisation of the initially formed alkenes was delayed, except with III, where for the same reason some Z-E isomerisation started early.
364 |
CHAPTER 8 |
Figure 8.2. Reaction scheme for the hydrogenation of 3,4-dimethyl-1,2-pentadiene: configurations of the adsorbed reactant are shown both as π- and di-σ -structures (alkenes only isomerise after the reactant has disappeared).
Favoured orientations of I at the surface and the products to which it gives rise on Z-addition of two hydrogen atoms are shown in Figure 8.2. The structure in which the least substituted double bond is adsorbed is preferred, but only by a factor of about two over the alternative: similar results were obtained with II and III, so the size and number of the substituents seems to make little difference. This is the more easily understood when it is recalled that even in the π-adsorbed state planarity is modified, a tendency that is even more marked if the di-σ-adsorbed state is envisaged, although this is unlikely with palladium. However, the way in which the first-formed alkenes are created by Z-addition of hydrogen is perhaps more easily visualised if the reactant structures are written as di-σ states (see Figure 8.2). Isomerisation of the initial products occurs when they can regain access to the surface, giving the stabler structures in which the number of substituents is maximised and their steric repulsions minimised. A very dramatic increase in regioselectivity occurs if a substituent is a functional group that is capable of anchoring the molecule to the surface, since H2C C
C CH (COOCH3) is reduced with 100% selectivity to CH3−CH
CH (COOCH3) is reduced with 100% selectivity to CH3−CH CH(COOCH3).42
CH(COOCH3).42
Allenic dienes having four different substituents possess chirality, as noted above: the various possible modes of adsorption and the structures of products resulting from their hydrogenation have been explored.42
HYDROGENATION OF ALKADIENES AND POLY-ENES |
365 |
Figure 8.3. The half-hydrogenated state of cycle-1,2-nonadiene shown in plan as (A) a π3 structure and (B) a πσ structure.
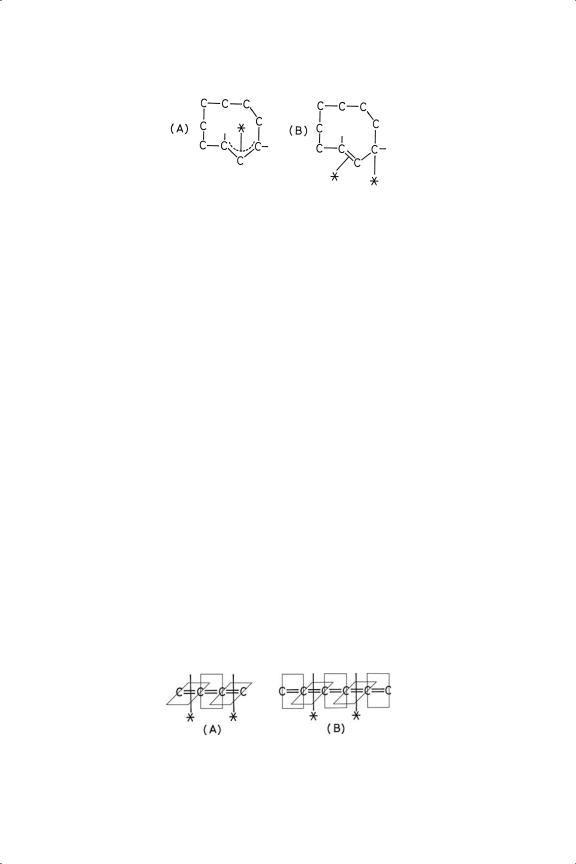
If an alicyclic ring be large enough, it can accommodate a 1,2-diene group, but the ring must have at least nine carbon atoms for this to be possible. Hydrogenation of cyclo-1, 2-nonadiene and -decadiene over Pd/C poisoned with pyridine afforded12,43 the corresponding Z-cycloalkene exclusively or predominantly, although the E-isomers were converted to Z-isomers, the latter being stabler due to the constraints imposed by the ring. However E-isomers constituted respectively 17 and 32% of the initial products, and it was argued that, since application of the foregoing principles should lead only to Z-isomers, the reaction must have proceeded through a π-allylic half-hydrogenated state, the stablest configuration of which (Figure 8.3) permits the two isomers to be formed with equal probability. However, the planar πσ-structure (Figure 8.3B) would answer just as well.
8.2.3. Hydrogenation of Cumulenes
It is possible for more than three carbon atoms to be connected by double bonds: compounds are known in which four or six atoms are so conjoined, and are known as cumulenes. Their probable modes of chemisorption are as shown in Figure 8.4, because the tetra-phenyl compounds have been hydrogenated on the Lindlar catalyst (Pd-Pb/CaCO3) to respectively the 1,3-diene or to the 1,3(Z ), 5-triene.2,4
8.3. HYDROGENATION OF 1,3-BUTADIENE
8.3.1. General Characteristics of Butadiene Hydrogenation3,22,44
The principal parameters that characterise this reaction are (i) the selectivity to the sum of all three butenes Stot, and (ii) the fraction of the butenes that
Figure 8.4. Di-π chemisorbed states of (A) C4 - and (B) C6 -cumulenes.

366 |
CHAPTER 8 |
each isomer constitutes (S1, SZ2 and SE2). Over many of the metals of Groups 8 to 10, Stot is close to unity and (as with 1,2-butadiene) the composition of the isomers is unchanged until the reactant has been consumed: it is therefore more strongly adsorbed than the products. In harmony with this, orders of reaction are frequently about zero in butadiene and first in hydrogen. On platinum, however, Stot is usually much less than unity. The composition of the butenes varies widely: sometimes the main product is 1-butene, with significant E -2-butene but little Z -2-butene (Type A behaviour), but occasionally the E/Z ratio is very high ( 8; Type B behaviour); intermediate situations are not unknown. Mechanisms attempting to account for these situations, and for the results of experiments using deuterium, will be considered in Section 8.3.6. Activation energies are typically about 40 kJ mol−1, a value that is typical for a true activation energy where the surface composition does not change much with temperature; this is consistent with the observed near-zero orders in butdiene.
Since the reaction was first studied in depth, in the early 1960s, it has proved an attractive means of exploring how catalyst structure and composition determine behaviour: unlike, say, the hydrogenation of ethyne, where there is only a single selectivity to be measured, butadiene hydrogen affords the additional flexibility provided by the formation of the three butenes, and it was hoped, with some justification as it turned out, that these would reveal additional depths of mechanistic understanding. The reaction often shows a lack of response of rate and of selectivities to particle size, and is therefore deemed to be structure-insensitive. Effects attributable to other variables such as deliberate or accidental additives or to the nature of the support can be however quite marked. The industrial relevance of the reaction,8 noted above, has led to a number of works in which bimetallic catalysts have been used, because it has long been know that the good selectivity shown by palladium could be made excellent by the addition of another metal, especially one from Group 11. This work is summarised in Section 8.3.7.
We begin by looking at the limited work carried out on single crystals (Sections 8.3.2 and 8.3.3), and then more to supported metals (Section 8.3.4) and finally to other unsupported forms (Section 8.3.5). Larger linear, branched and cyclic molecules containing two or more conjugated double bonds are dealt with in Section 8.4.
8.3.2. Chemisorbed States of 1, 3-Butadiene45,46
This molecule has not enjoyed the popularity of ethene and ethyne, but it has been subjected to a number of studies on supported metals:45 unfortunately these do not lead to any very firm conclusions, partly because polymerisation or decomposition intervene to cloud the picture. Two studies on single crystal surfaces do however provide firmer and quite important conclusions. On Pt(111) at 9546 or