

Metal-Catalysed Reactions of Hydrocarbons / 11-Hydrogenation of Small Alicyclic Rings
.pdf11
HYDROGENATION OF SMALL
ALICYCLIC RINGS
PREFACE
Cyclopropane is a fascinating molecule: although its carbon atoms are linked by what are formally single C––C bonds, it partakes some of the properties of an alkene. It is quite easily hydrogenated, but much less strongly adsorbed than alkenes. As with benzene, its bonding defies simple description, and one might say it is ‘neither fish, flesh, fowl or good red herring’. Although only of small practical value, its hydrogenation and particularly that of its alkyl-substituted derivatives have been quite widely used as a way of assessing aspects of catalyst structure and composition that determine activity and specificity: but it needs to be said that the interpretation of what is observed falls short of what might have been hoped for.
The convention is adopted here of calling the processes of converting the cyclopropane and cyclobutane rings to the corresponding alkanes hydrogenation, and the breaking of further C––C bonds when it occurs (as it does simultaneously at higher temperature) hydrogenolysis. Cyclobutane is more like an alkane and less like an alkene than is cyclopropane; however, further enlargement of the ring to cyclopentane gives a molecule that is undoubtedly a cyclic alkane, although as we shall see (Chapter 14) this too has some unusual properties Alkene character diminishes with increasing ring size, rather like the Cheshire cat, which slowly disappeared, starting from its tail, until only the smile was left.
11.1. INTRODUCTION
It might at first sight seem unlikely that a stable molecule could be formed by joining three methylene groups as in a triangle, since distortion of hybrid sp3 orbitals to allow a C––C––C bond angle of 60◦ would appear impossible. However,
473

474 CHAPTER 11
TABLE 11.1. Some Physical and Structural Properties of Small Alicyclic Rings Cx H2x1
|
L(C––C)/nm |
D(C––C)/kJ mol−1 |
− |
f |
− |
HH /kJ mol−1 |
|
x |
|
H |
—O /kJ mol−1 |
|
|||
3 |
0.150 |
238 |
|
|
53.3 |
|
157 |
4 |
0.156 |
293 |
|
|
27.7 |
|
153 |
5 |
0.154 |
385 |
|
|
77.3 |
|
69 |
6 |
0.154 |
397 |
|
|
123.2 |
|
44 |
|
|
||||||
L(C––C) = bond length; D(C––C) = bond dissociation energy; − |
Hf —O = standard heat of formation; − HH = heat |
||||||
of hydrogenation to the corresponding alkane. |
|
|
|
|
|
||
cyclopropane is a comparatively stable molecule, and the fact of its existence has taxed the subtlety of the minds of theoreticians for more than half a century. The strain imposed by its geometry gives it some of the characteristics of an alkene, because it undergoes addition with halogens and with hydrogen, this latter process being our particular concern. Its heat of hydrogenation to propane is greater than that of ethene to ethane (Table 11.1); its C––C bond length (0.152 nm) is shorter than that in ethane, but the C––C bond dissociation energy is much less than that in the larger alicyclic molecules.1 It enters into conjugation with C C bonds, but is unable to transmit it, and in many other respects its properties are intermediate between those of alkenes and alkanes.2 In a word we have a mystery wrapped in an enigma.
C bonds, but is unable to transmit it, and in many other respects its properties are intermediate between those of alkenes and alkanes.2 In a word we have a mystery wrapped in an enigma.
The several attempts3 that have been made to find theoretical frameworks for understanding these unique properties need not delay us for long. The earliest picture was that proposed by Donald Walsh in 1949:4 he suggested that the C––C bonds are formed by the intra-annular overlap of one of the sp2-hybridised orbitals of each carbon atom with three p orbitals. This creates a concentration of charge in the centre of the ring, and leads to one concept of how the molecule can chemisorb (see below). A valence-bond approach5,6 led to a “bent-bond” model, in which hybridised sp4.12-orbital lobes formed angles of 104◦, and overlapped outside the equilateral triangle. One study5 aimed to show that these two approaches were compatible. An ab initio SCF-MO calculation6 did not support Walsh’s model, but concluded that the molecule’s reactivity was due to a relatively low-lying excited σ -orbital, in contrast to alkenes, where the analogous orbital has π -character. A recent survey3 concludes that the orbitals directed to the hydrogen atoms have sp2 character, while those to adjacent carbon atoms are approximately sp5, the extra p character relieving some of the strain that would feature in sp3 hybridisation. The electron density is then directed away from the ring at an angle of 21◦ to the line joining the carbon nuclei, confirming the ‘bent-bond’ model noted above. Since cyclopropanes can be synthesised by insertion of a methylene diradical (e.g. from diazomethane) into a C C bond, it has also been possible to regard the bonding as being the result of resonance between the three possible canonical forms (Figure 11.1). The reactivity of and the bonding in the C3 ring are often discussed in organic
C bond, it has also been possible to regard the bonding as being the result of resonance between the three possible canonical forms (Figure 11.1). The reactivity of and the bonding in the C3 ring are often discussed in organic

HYDROGENATION OF SMALL ALICYCLIC RINGS |
475 |
Figure 11.1. Representation of cyclopropane as a resonance hybrid of methylene and ethene.
chemistry texts; for recent experimental and theoretical studies, see references 7 and 8.
Alkenic unsaturation in conjunction with the cyclopropane ring has interesting theoretical and practical effects. Methylenecyclopropane is perhaps better formulated as trimethylenemethane;9 its hydrogenation2 and that of derivatives10 has been examined (Section 11.4). Cyclopropene sounds as if it would be very highly strained, but its methyl derivatives have apparently been made and hydrogenated11 (see also Section 11.4).
Not much is known about the chemisorption of cyclopropane.12 Its heat of adsorption on platinum film has been measured13 (197 kJ mol−1), but like other hydrocarbons is suffers variety of fragmentation processes on clean metal surfaces;14−17 self-hydrogenation leads only to propane on palladium and platinum, while on other metals (Rh, Ni, Mo, Fe), more active in hydrogenolysis, methane and ethane are also formed.18 Reactions in the absence of hydrogen, brought about only by hydrogen atoms derived from the reactant, run parallel to those in the presence of hydrogen. Three forms have been suggested as intermediates in the hydrogenation to propane (Fig. 11.2). Structure A shows the molecule lying flat on the surface, bonded to a metal atom by interaction of delocalised electrons within the ring with vacant orbitals on the metal. UHV studies provide some evidence for this form at low temperatures on single crystals,14 and it harmonises with Walsh’s model, but no detailed description of the bonding has been offered. Structure B is a dissociatively-adsorbed state typically shown by alkanes, but it has been dismissed because reaction with deuterium only very rarely leads to cyclopropanes-dx , and then usually just in small amounts (Section 11.2.2). Structure C is clearly destined to lead to propane, and it could arise through A as a transitory state. Many other species containing more or fewer hydrogen atoms have been advanced as intermediates in the degradation, hydrogenation or hydrogenolysis, and we shall note some
A B C
Figure 11.2. Possible adsorbed states of cyclopropane.
476 |
CHAPTER 11 |
of them presently. The kinetics of hydrogenation (Section 11.2) do however show clearly that cyclopropane is usually much less strongly adsorbed than hydrogen.
One of the significant features of the reaction of cyclopropane with hydrogen is that on metals active for hydrogenolysis, i.e. all the metals of Groups 8 to 10 excepting palladium and platinum, fragmentation to methane and ethane takes place at much lower temperatures than would occur with propane. Ethene and propene are also observed under certain conditions.15
Much interest has been shown in the regiospecificity of the hydrogenation of alkyl-substituted cyclopropanes (Section 11.3). In general it is the least obstructed C––C bond that is broken most easily, so for example methylcyclopropane gives chiefly isobutane, but the alternative mode giving n-butane has a higher activation energy, and thus becomes more important as temperature is raised.2,19 This reaction has often been deployed in attempts to provide a more detailed view of the structure of a catalyst’s surface19,20 (Section 11.3.1), but it has to be said that the results are somewhat disappointing. While 1,2-dimethycyclopropane gives mainly 2-methylpentane, when adjacent substituents form part of another cycle the intervening C––C bond is strained, and breaks first. This permits one of the few synthetically useful reactions of cyclopropane to be effected: for example, methylene insertion into cyclohexene gives bicyclo[4.1.0]heptane, which is converted by hydrogenation to cycloheptane.
The cyclopropane ring can exist in a variety of environments:1,10,11 among the more interesting structures the hydrogenation of which has been investigated are spiropentane (bicyclo[2.2.0]pentane), bicyclopropyl, cyclopropylmethanes ((C3H5)x CH4−x , x = 1 to 4), nortricyclene and phenylcyclopropane (Figure 11.3; see Section 11.3.3). Fusion of a C C double bond directly onto the cyclopropyl ring entirely changes the mode of ring breaking, so that a bond adjacent to the
C double bond directly onto the cyclopropyl ring entirely changes the mode of ring breaking, so that a bond adjacent to the
Figure 11.3. Structures of molecules containing the cyclopropane ring.

HYDROGENATION OF SMALL ALICYCLIC RINGS |
477 |
point of substitution is now the most reactive: the same effect is produces by an ethenyl substituent (Section 11.4).
The cyclobutane ring behaves much more like an alkane:1 its hydrogenation requires significantly higher temperature than does cyclopropane (Section 11.4). The ring is puckered, with a dihedral angle of 20◦, and the C––C bonds are longer than those in ethane or the larger alicyclic molecules; other relevant facts are given in Table 11.1. Its alkyland methylenesubstituted derivatives have also enjoyed much attention, presumably for the same reason as the corresponding cyclopropanes.
11.2.HYDROGENATION AND HYDROGENOLYSIS OF CYCLOPROPANE
11.2.1. Kinetics
There have been numerous studies of the kinetics of the reaction of hydrogen with cyclopropane, but some record only activation energies and not orders of reaction. Some distinguish between hydrogenation to propane and hydrogenolysis to methane and ethane, the kinetics of these two types of process being quite different (see Tables 11.2 and 11.3): many however have contented themselves with measuring parameters of the overall reaction, or had to of necessity because the means of analysis were lacking. We focus first on hydrogenation.
The difference between cyclopropane and alkenes on the one hand and alkanes on the other is nowhere more clearly seen than in the kinetics of their reactions with hydrogen and in the relative activities of metals for those reactions. The ranking of metals activities for cyclopropane hydrogenation,21−23 namely,
Rh > Ni > Pt > Pd > Ir > Ru Os > Co
is quite different from that for alkane hydrogenolysis, but depends on the surface cleanliness temperature and reactant concentrations at which the rates were measured. The earliest kinetic work showed that on the Group 10 metals the order in hydrogen was zero and in cyclopropane first; the former clearly saturated the surface and the latter was at most weakly adsorbed.24 Extension of this work and the use of other metals refined this simple picture. Rates frequently passed through maxima as hydrogen pressure was raised, the position of the maximum increasing with temperature, and the order above the maximum becoming less negative.2 The negative values quoted in Table 11.2 were measured at 323 K. Orders in cyclopropane became more positive as the fixed hydrogen pressure was raised, and with temperature at constant hydrogen pressure. This behaviour is typical for the competitive adsorption of the reactants, and the application of Langmuir-Hinshelwood

478 CHAPTER 11
TABLE 11.2. Kinetics of the Hydrogenation of Cyclopropane (r PH x Phc y )
Metal |
Form |
E / kJ mol−1 |
x |
y |
|
T /K |
Reference |
Fe |
a |
56 |
−1 |
1 |
|
373 |
39 |
|
|
||||||
Co |
Powder |
45 |
−0.1 |
0.7 |
|
394 |
23 |
Ni |
Powder |
54b |
−0.1 |
0.8 |
|
300 |
23 |
Ni |
Film |
31 |
−0.1 |
0.6 |
|
250 |
27 |
Ni |
/Pumice |
44 |
0 |
1 |
|
443 |
24 |
Ni |
/SiO2 |
54 |
−0.1 |
0.8 |
|
300 |
87 |
Ni |
/SiO2 –A12 O3 |
58 |
−0.2 |
0.6 |
|
366 |
87 |
Ru |
(0001) |
26 |
0.75 |
0 |
|
450 |
14 |
Ru |
(1120) |
20 |
0.9 |
0 |
|
450 |
14 |
Rh |
Pumice |
42b |
−0.36 |
0.4 |
|
323 |
2 |
Pd |
Film |
61 |
−0.9 |
0.1 |
|
250 |
27 |
Pd |
/Pumice |
34 |
0 |
1 |
|
395 |
24 |
Pd |
/Pumice |
— |
−0.8 |
c |
|
323 |
29 |
0.4c |
|
||||||
Pd |
/Pumice |
42 |
−0.45 |
0.4 |
|
323 |
2 |
Ir |
(111) |
41 |
1 → −0.5 |
+ve |
|
450 |
15 |
Ir |
(110)(1 × 2) |
36 |
1 |
0c |
|
425 |
15 |
Ir |
/Pumice |
48 |
−0.2 |
0.4 |
c |
323 |
2 |
Ir |
/Pumice |
41 |
0 |
0.53 |
|
323 |
29 |
Pt |
Powder |
51b |
−0.5 |
1 |
|
352 |
23 |
Pt |
Film |
46 |
−0.2 |
0.2 |
|
223 |
27 |
Pt |
/Pumice |
37 |
0 |
1 |
|
356 |
24 |
Pt |
/Pumice |
33 |
−0.35 |
0.9c |
|
473 |
28 |
Pt |
/Pumice |
36 |
−0.3 → −0.6 |
0.9 |
|
323 |
2 |
Cu |
Powder |
46 |
−0.1 |
0.6 |
|
419 |
23 |
a Reduced magnetite with added K. b Reaction with D2.
c Measured using the H2 pressure giving maximum rate.
formulism based on a rate-controlling reaction of a hydrogen atom with an adsorbed cyclopropane led to estimates of the adsorption coefficients for hydrogen, which decreased, as they should, with increasing temperature and indeed as the order suggested they would. However, the values depended very much on the method of preparation of the catalyst, and since dispersions were not at that time measured it is likely that their variation was at least partly responsible. Measurement of the hydrogen order for this reaction might be developed into a simple way for estimating metal dispersion.
Other forms of the Langmuir-Hinshelwood method have been tried. Orders in hydrogen more negative than −0.4 seemed to require the participation of a chemisorbed hydrogen molecule,2 while an equation based on a slow reaction of a hydrogen atom with a propyl radical worked for a Pt/SiO2, but not for EUROPT-1 (6% Pt/SiO2).25,26 Other workers have failed to extract maximum significance from their results by expressing them as exponents of reactant pressures, changing from positive to negative in ways not well explained. Table 11.2 shows a selection

HYDROGENATION OF SMALL ALICYCLIC RINGS |
479 |
of reaction orders expressed in this way: for supported metals,2 powders23 and films,27 hydrogen orders are zero or slightly negative, while those for cyclopropane are generally positive (0.4 < y < 1).2,28,29 Values of activation energies shown in Table 11.2, and others not shown there,16,18,22,30−33 are mostly in the range 45 ± 10 kJ mol−1. If values close to the mean were found under conditions where the surface concentrations were not changing much, 45 kJ mol−1might be regarded as the ‘true’ activation energy. Exceptions to these generalisations are shown by iron catalysts (where hydrogenolysis intrudes on powder16 and film19) and by palladium;22,27 in these cases, hydrogen appears to be much more strongly chemisorbed than cyclopropane. Ruthenium catalysts have shown both higher34 and lower14,33,34 values. Quite different behaviour has been shown by ruthenium single crystal surfaces14 and by Ir(110)(1 × 2);15 positive hydrogen orders and zero cyclopropane orders were observed, and in the former case activation energies were unusually low (20–26 kJ mol−1). These orders suggest that these surfaces are monopolised by cyclopropane molecules, or species derived from them.
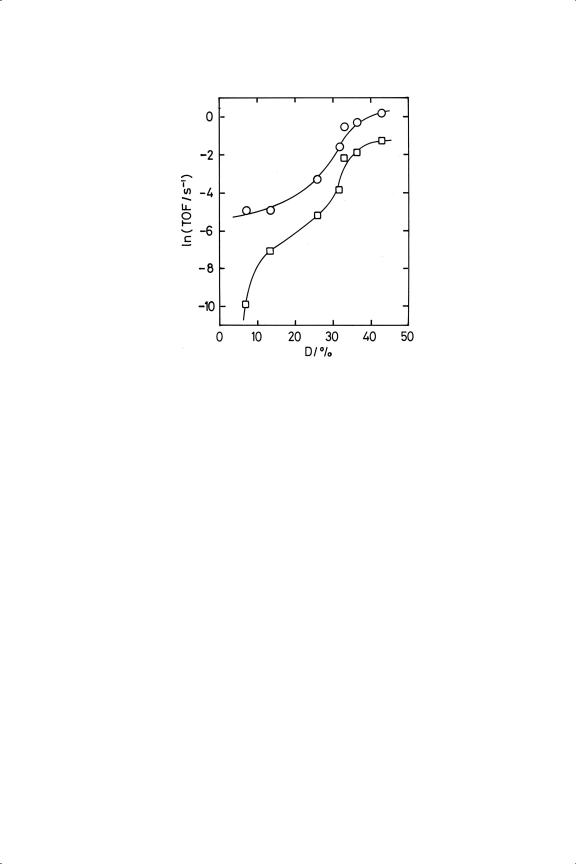
This virtual constancy of activation energy as metal, its form, and method of preparation are varied leads us naturally to consider the hydrogenation reaction’s structure sensitivity. It was probably the first hydrocarbon reaction to be declared structure-insensitive, by the lack of dependence of TOF on particle size, support and physical form:31,35,36 but this was for platinum, and later work with Ni/SiO237 showed maximum activity and minimum activation energy at a particle size of about 1 nm. Even more dramatic variations were shown by variously supported ruthenium catalysts,33 where those made from the trichloride had TOFs that increased by 105 as dispersion increased in the range 7 to 43% (Figure 11.4): activation energies between 24 and 49 kJ mol−1 showed no consistent size-dependence. Structure-sensitivity (or insensitivity) is therefore a function of the whole catalytic system, and not just of the reaction. It is difficult to generalise on such limited observations, but it seems likely that sensitivity is associated with the stronger adsorption of cyclopropane-derived species than hydrogen, and with propensity of metals to engage in simultaneous hydrogenolysis; indeed there may be a causal connection between these two occurrences (see later).
Ru/TiO2 catalysts have unusual character. On first reduction (HTR1) of RuCl3/TiO2, catalysts of only moderate activity were obtained, because they are somewhat poisoned by chloride ion and partially in the SMSI state (Section 3.3.5). Oxidation followed by low-temperature reduction (O/LTR) removed chloride ion and greatly increased particle size; much higher activities for cyclopropane hydrogenation (and alkane hydrogenolysis, see Chapter 13) were obtained.34 A second high-temperature reduction (HTR2) converted catalysts very largely into the SMSI state, with loss of much of their activity. However, in contrast to the situation with alkanes, with cyclopropane the most active catalyst has the highest activation energy. Hydrogenation selectivity decreased in the sequence HTR2 > HTR1 > O/LTR, indicating that (not surprisingly) hydrogenolysis requires a larger ensemble
480 |
CHAPTER 11 |
Figure 11.4. TOFs for the hydrogenation (circles) and hydrogenolysis (squares) of cyclopropane on variously supported ruthenium catalysts of different dispersion.33
of free atoms than does hydrogenation. Reduction of Rh/La2O3 at 673 K or above has led to most of the activity for cyclopropane hydrogenation being lost; Rh/SiO2 was unaffected.38
On metals of moderate to high activity for hydrogenolysis, i.e. on all metals of Groups 8 to 10, except palladium and platinum, the splitting of a second C––C bond accompanies the breaking of the first, methane and ethane being the products.14−16,18,22,23,27,37 Some ethene may also be formed, but complete conversion to methane requires high temperatures: methane/ethane ratios greater than unity have also been found particularly with large (>5 nm) ruthenium particles,33 and the activation energy was then very high (94 kJ mol−1). Activation energies for hydrogenolysis were in general somewhat larger than for hydrogenation,14,23,33,34,37, so that the former became more important as temperature was raised. With supported metals, orders of reaction for the two processes did not differ greatly, orders for hydrogenolysis tending to be more negative in hydrogen and less positive in cyclopropane (Table 11.3). The situation was clearest with the single-crystal studies: on ruthenium surfaces, the highest selectivities to propane were found at low hydrogen/cyclopropane ratios,14 but on Ir(111) more extensive cracking occurred at low hydrogen pressure.15 Hydrogenolysis was also structure-sensitive on nickel37 and ruthenium33 catalysts; in the latter case, selectivity to propane improved with decreasing dispersion (Figure 11.4). This may appear to conflict with the consequences of inducing SMSI with Ru/TiO2 catalysts,

HYDROGENATION OF SMALL ALICYCLIC RINGS |
481 |
TABLE 11.3. Kinetics of the Hydrogenolysis of Cyclopropane (r PH x Phc y )
Metal |
Form |
E /kJ mol−1 |
x |
y |
T /K |
Reference |
Co |
Powder |
78 |
−0.2 |
0.6 |
394 |
23 |
Ni |
Powder |
67 |
−0.3 |
0.4 |
300 |
23 |
Ni |
/SiO2 |
67 |
−0.3 |
0.4 |
300 |
87 |
Ni |
/SiO2 -A12 O3 |
55 |
0.5 |
0.6 |
366 |
87 |
Ru |
(0001) |
43 |
1.8 |
−0.5 |
450 |
14 |
Ru |
(1120) |
43 |
1.8 |
−0.6 |
450 |
14 |
Ir |
(111) |
111 |
+ve. |
0 |
450 |
15 |
Ir |
(110)-(1×2) |
96 |
0 |
0 |
425 |
15 |
but the effects cannot be strictly comparable. Extensive fragmentation of cyclopropane occurred on iron catalysts.16,27,39,
Little work has been reported on the use of bimetallic catalysts.32,40,41 Addition of copper to nickel as powder lowered the overall rate, without major change to the propane selectivity. Activation energies varied irrationally between 40 and 93 kJ mol−1.
11.2.2. The Reaction of Cyclopropane with Deuterium42
Early work using Pt/pumice showed28 that the main initial products at 273 K were propanes-dx (x = 2 to 4), but unless a large excess of deuterium was used its hydrogen content rose with increasing conversion, and mean deuterium number (M) of the propane fell. The value of M increased from about four to six as temperature increased to 473 K; this was due to a marked increase in the amounts of propane-d7 and -d8 formed, their activation energies being significantly higher than those for the lighter molecules. At constant temperatures (273 and 473 K), however, product distributions were independent of the deuterium/cyclopropane ratio. Thus at 473 K, propanes-d7 and -d8 were the major products, but a secondary maximum appeared at propane-d4. Later work29 confirmed and extended these measurements, and distributions of propanes-dx were modelled by random extraction of atoms from two ‘pools’. Pool A had a deuterium content that rose from 90 to 96% and constituted 23 to 64% of the whole between 323 and 473 K, while pool B had 45–50% deuterium and decreased from 71 to 33% of the whole; a small amount of ‘direct addition’ to propane-d2 was also suggested. Similar studies with pumice-supported palladium, rhodium and iridium gave broadly similar results, but yields of propanes-d7 and -d8 were higher (Pd, 80%; Rh, 77%; Ir, 80%) and essentially independent of reaction conditions. The same mode of interpretation was adopted.
It very soon became clear that the products of the reaction of cyclopropane with deuterium could be explained by supposing a low concentration of propyl radicals and a high concentration of deuterium atoms on the metal surfaces, their

482 |
CHAPTER 11 |
interactions being similar to those experienced by alkanes, with multiple exchange predominating. Parallel measurements28,29 with propane confirmed this picture, although there were significant differences, as activation energies were higher and activities lower, and a stepwise exchange was also noted. These changes were due to the greater ease of chemisorbing cyclopropane, and to the fact that the opening step created structure C (Figure 11.2) and not a propyl radical. Propyl radicals were also formed by dissociation of 1- and 2-chloropropane, and behaved similarly.29,43
On films of the metals of Group 10, rhodium, iron and tungsten, the cyclopropane-deuterium reaction gave product distributions in line with those found with the pumice-supported metals where comparison is possible; nickel and particularly iron gave much multiply-exchange propanes.27 The kinetic parameters are given in Table 11.2.
Very slight exchange of the hydrogens in cyclopropane was noted on Rh/pumice and Ir/pumice;29 it appeared to be substantial on rhodium film at 173 K,44 suggesting that it is a low activation energy process. It also occurred significantly on tungsten film.27
More recently the reaction has been examined over silica-supported rhodium, palladium, iridium and platinum, with the help of NMR spectroscopy.45 The results were much in line with those already mentioned, the amounts of propane-d8 increasing in the sequence: Pt < Ir < Rh < Pd. The propane-d2 was not surprisingly mainly propane-1,3-d2. A new feature was observed with Ir/Al2O3; the support made a significant contribution to the overall reaction, and this necessitated a revision of a previous interpretation of the activity of this metal in alkane exchange.46
11.2.3. Reaction Mechanisms
Discussion of the mechanisms involved in the reactions of cyclopropane with hydrogen and deuterium is relatively straightforward because of the evident similarity if not identity of unit steps with those encountered in the reactions of alkanes (Chapters 6, 13 and 14). This has not however inhibited scientists from suggesting a variety of possible intermediate adsorbed species.
The exchange process can be swiftly dismissed because of its small importance. Its simplest formulation involves the reversible dissociative chemisorption of the cyclopropane molecule as structure B (Figure 11.2). However, the reactivity of the C––H bond is probably not that much different from the bond in alkanes, while cyclopropane exchange appears to go best at low temperature, and has a low (and possibly even negative) activation energy. An alternative route is therefore through the undissociated structure A, where exchange occurs as a simple displacement (Scheme 11.1). There is no kinetic information to assist us.
The kinetics of the hydrogenation clearly point to competitive adsorption of the reactants, the sign before the hydrogen order and the magnitude of the