

Metal-Catalysed Reactions of Hydrocarbons / 07-Hydrogenation of Alkenes and Related Processes
.pdf7
HYDROGENATION OF ALKENES AND RELATED PROCESSES
PREFACE
It is now time to consider a catalytic reaction in which the product is distinctly different from the reactant hydrocarbon – the hydrogenation of an alkene (they used to be called ‘olefins’; some people do still call them that). As is typically the case, the first member of the series – ethene – has received more than its fair share of attention; this is understandable up to a point, because when it reacts with hydrogen ethane is always the main product, and usually the only one. Also it cannot compete with ethene for the surface, a fact that simplifies the kinetics somewhat. However the reaction with deuterium reveals a level of complexity unsuspected when only hydrogen was used, and the same Horiuti-Polanyi mechanism, which so satisfactorily explains what occurs, also accounts easily for the occurrence of reactions shown by the higher linear alkenes, in which the location and stereochemistry of the double bond changes concomitantly with the process of hydrogen addition. Related stereochemical subtleties are shown in the hydrogenation of substituted cycloalkenes. Just about every catalytic chemist who worked with metals will at some time have hydrogenated ethene, as its apparent simplicity and ease of performance commends it as an attractive way of assessing whether and how the physicochemical nature of the catalyst determines its activity. Many papers are content to record just that: but there have also been profound studies of its mechanism on a limited range of catalysts, which result in this being one of the best understood of catalysed hydrocarbon transformations.
291

292 |
CHAPTER 7 |
7.1. INTRODUCTION1–6
The metal-catalysed hydrogenation of ethene was discovered by Paul Sabatier in the early years of the last century, after which it received little attention until the 1930s when the application of deuterium started to reveal the inner complexities of this apparently simple reaction.7−9 These studies and those that followed (Section 7.2.4) demonstrated that the reaction is much more than the simple addition of a molecule of hydrogen or of two hydrogen atoms to the carbon-carbon double bond, but this has not prevented its use as an easy way of seeing how changes to the nature of catalysts affect their catalytic behaviour. However most of this extensive work, and the numbers of papers describing it must run into hundreds, records only the conversion or rate under a single set of operating conditions, and, because the response to changes in these variable has not been explored, it is of limited value: nevertheless we will have to note the conclusions reached by using catalysts having different particle sizes (Section 7.2.3). I fear that this apparent want of extended and systematic examination of the dependence of catalytic power on chemical composition and physical structure will be a recurring litany, because it can be said—and it needs to be said—of every class of reaction. I just hope it will not bore the reader.
The attraction of the reaction when performed with hydrogen lies in the singular nature of the final product, because with ethene, and indeed the higher linear alkenes also, the activity of many metals is such that reaction occurs in a range of temperature where the position of equilibrium greatly favours the alkane. Ethene hydrogenation can be followed on metal films even at 173 K. It is only with cyclohexenes, where further dehydrogenation is facilitated by the resonance stabilisation of the aromatic product, that products other than the alkane are observed at moderate temperature. The only other slight complication is that homologation is found in some circumstances (Section 7.3.1). Rates are usually faster than those of alkadienes and alkynes (see Chapters 8 and 9) because the reactive form is less strongly adsorbed; a ‘volcano’ relation between reactivity and adsorption strength has been proposed.10
Alkene hydrogenation is significantly exothermic, and it is not always easy to keep the catalyst isothermal except at low rates. Heats of hydrogenation for a number of alkenes were measured many years ago (Table 7.1),2 the use of a catalyst ensuring that calorimetry could be conducted at ambient temperature. The values are similar to but perhaps more accurate than those derived from heats of combustion, where subtraction of two large numbers is involved; they reflect the extents to which the π electrons interact with the electrons in the C––H bonds by hyperconjugation. This electron delocalisation is also reflected in the relative stabilities of alkene complexes with Ag+ cations.2
Measurements of the specific rate of ethene hydrogenation made under fixed conditions on catalysts of the same physical form reveal a systematic variation

HYDROGENATION OF ALKENES AND RELATED PROCESSES |
293 |
TABLE 7.1. Heats of Hydrogenation of Alkenes (kJ mol−1 )2,64,249,250
Linear Alkenes |
− HH |
Branched Alkanes |
− HH |
Cycloalkenes |
− HH |
Ethene |
137 |
2-Methyl-propene |
119 |
Cyclopentene |
109 |
Propene |
124 |
2-Methyl-2-butene |
112 |
Cyclohexene |
113 |
1-Butene |
126 |
2,3-Dimethyl-2-butene |
111 |
Cycloheptene |
108 |
E-2-Butene |
115 |
4-Methyl-1-pentene |
121 |
Cyclo-octene |
96 |
Z-2-Butene |
119 |
2-Methyl-l-pentene |
112 |
1,2–Dimethylcyclo- |
112.5 (Z) |
|
|
|
|
hexene |
120.5 (E) |
1-Pentene |
109 |
4-Methyl-E-2-pentene |
110 |
|
|
E-2-Pentene |
114 |
|
|
|
|
Z-2-Pentene |
118 |
|
|
|
|
|
|
|
|
|
|
Calculated by DFT by Dr. E.L. Short for conversion respectively to Z- and E-dimethyl-cyclohexane.
in activity with the metals’ electronic structure,2 and hence with the strengths of adsorption of both of the reactants, which according to Beeck’s results11,12 vary in the same way with Periodic Group number: this is as expected if the slow step requires the breaking of M––H and M––C bonds. Thus in accordance with the Sabatier or Volcano Principle, the activity maximum is located where the best compromise between coverage and bond strength is to be found (Figure 7.1). This occurs at Group 9 or Group 10 (Ir is unaccountably 102 less active than Rh according to these old results), and the general level of activity of the base metals is less than that of the noble metals in Group 8 to 10.3 Measuring rates at constant reactant pressures and temperature is no guarantee that the number of reacting
Figure 7.1. Dependence of the activity of metals for ethene hydrogenation on Periodic Group number.2,3,11 ,First row metals; , second row metals; O, third row metals. Open points, condensed metal films; filled points, silica-supported metals.

294 |
CHAPTER 7 |
centres will be the same, and the lower activities of the base metals may simply reflect smaller areas of active surface resulting from their greater tendency to cause extensive disruption of the alkene.
Correlations between activity, adsorption strengths, and fundamental properties of the metals, both measured and derived, have however long been canvassed,12 and have pedagogic value for discussing the physical basis of catalytic activity, at least in this class of reaction.3 However the revelation of the more complex nature of the reaction by the use of deuterium in place of hydrogen13 provides an additional level for analysing these connections. It will therefore be helpful to anticipate the discussion of the results of isotopic tracer experiments (Section 7.2.4) by setting down the basic mechanisms and terminology at this point.
Numerous works have sought to define the mechanism of alkene hydrogenation, as if this were a universal truth, applicable in all circumstances. Our discussion (Section 5.3) teaches that a more catholic view is appropriate, and that we should regard the mechanistic scenario as a kind of pot containing all conceivable elementary steps, from which we withdraw such items and in such quantities as are requisite to account for the observations in any particular case. We must abandon the idea that a single mechanistic statement will be valid everywhere. The Horiuti-Polanyi mechanism, to which reference has already been made in the context of alkane exchange, therefore represents the simplest possible way of accounting for what happens when ethene reacts with deuterium. As we shall see, it has been necessary to elaborate the original idea: this does not however demean its value.
The early work on this reaction disclosed dilution of the deuterium content of the non-hydrocarbon reactant by hydrogen; this could only be explained by the simultaneous occurrence of an alkene exchange reaction,7,14−16 i.e.
C2H4 + D2 → C2H3D + HD |
(7.A) |
An inevitable consequence is the subsequent formation of more extensively exchanged ethene and of ethanes containing from zero to six deuterium atoms, but the relative rates of the exchange and addition vary widely with the metal and with operating conditions (Section 7.31). These observations are neatly explained by the Horiuti-Polanyi mechanism,8 which proposed a reversible stepwise addition of deuterium (or hydrogen) atoms, e.g.
|
|
|
|
− H |
C2H4D |
|
|
+ D |
+ H |
|
|||
C2H4 |
|
|
|
C2H4D |
|
|
|
|
|
+ D |
(7.B) |
||
|
|
|
||||
|
− D |
C2H4D2 |
|
|||
|
|
|
|
− D |
|
|

HYDROGENATION OF ALKENES AND RELATED PROCESSES |
295 |
In this scheme the attachment of the species to the surface is not specified, and in particular the structure of the adsorbed ethene is left open for future discussion, as is the provenance of the deuterium atoms, although it will be evident that dissociative adsorption of the deuterium molecule must occur in some way. A more quantitative treatment of this scheme is deferred to Section 7.33. On some surfaces an intermolecular exchange of isotopes can take place in the absence of hydrogen or deuterium, e.g.
C4H8 + C3D6 → C4H7D + C3HD5 |
(7.C) |
The reaction presumably needs atoms liberated by dissociative chemisorption of the alkenes with the accompanying formation of alkyl radicals.2,17
A note on terminology: the symbol D represents the 2H isotope; in the text, D is called a deuterium atom and D2 a deuterium molecule; the same goes for H and H2. Terms such as protium (for 1H) or dideuterium (for D2) are eschewed. The process giving alkane is described as addition or hydrogenation, irrespective of whether hydrogen or deuterium species are involved. The terms deuterogenation and deuteriumation, being hybrids with all the elegance of a mule, are not employed.
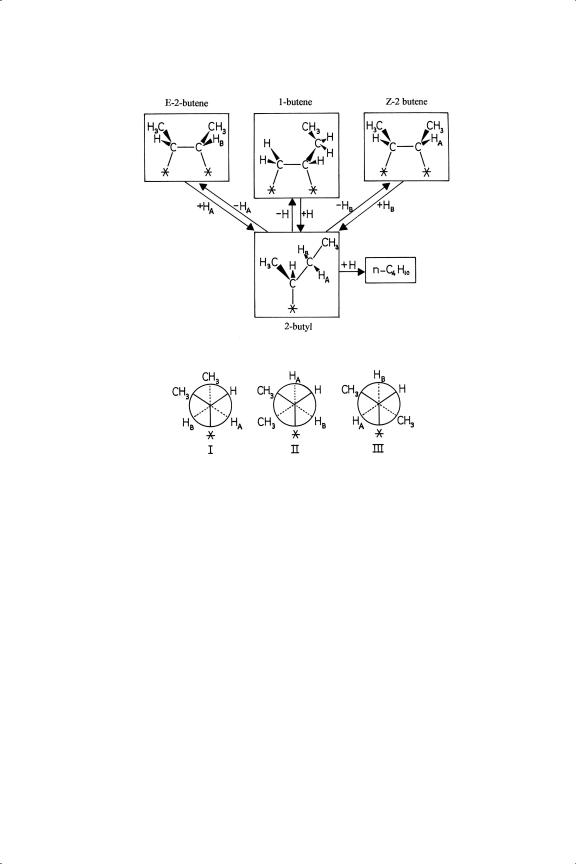
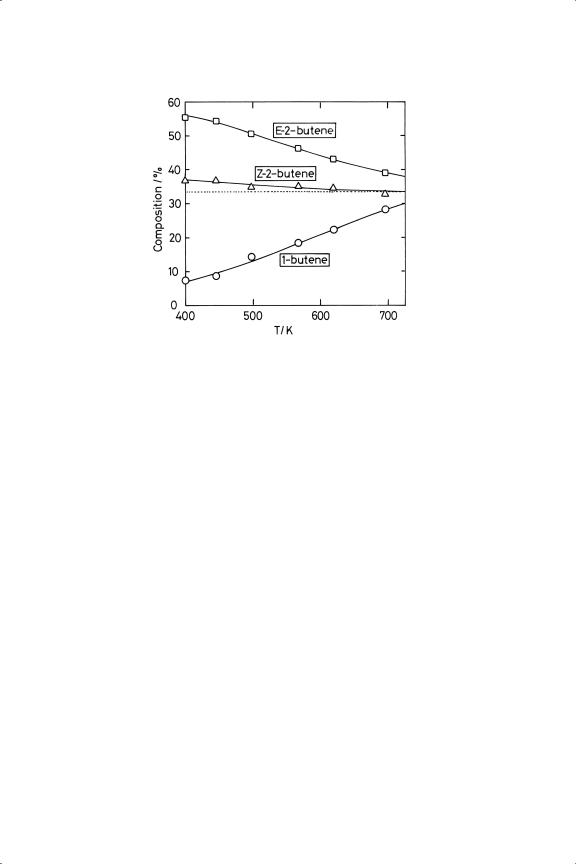
Recognition of the stepwise addition of atoms to a chemisorbed alkene and of the stability of the intermediate alkyl radical provided an immediate explanation for other processes that occur during hydrogenation of higher linear and cyclic alkenes. These are double-bond migration and cis-trans (or Z-E) isomerisation:2,6,18−20 these arise naturally because the atom removed in the alkyl reversal step is not necessarily that which was added, as is quite evident from the existence of alkene exchange. Once added, the atom is indistinguishable from other pre-existing atoms. Thus, when four carbon atoms are present, the reaction of 1-butene with hydrogen gives, besides n-butane, Z - and E -2-butenes, and the Z -isomer may transform to the E -isomer by rotation about the central bond of the 2-butyl radical (see Figure 7.2). The relative amounts of the various isomers formed do not correspond, at least initially, to those predicted by thermochemical considerations21,22 (Figure 7.3), because the proportions in which they return to the gas phase are determined by the energetics of their conformations in the adsorbed state and not by those in the free molecule.6 The isomer yields therefore contain mechanistic information and give valuable insights into the structures of adsorbed species. With catalysts that give much isomerisation, the amounts of the isomeric alkenes will approach or even reach their equilibrium values before all are hydrogenated (Sections 7.3 and 7.4); 3-methylcyclohexene will change to the 1-isomer, where the double bond stays because this is where it is most comfortable. Hydrogenation of disubstituted cyclohexenes has interesting stereochemical consequences
296 |
CHAPTER 7 |
Figure 7.2. Interconversion of n-butene isomers via the σ -adsorbed 2-butyl radical: conformation I leads to either Z - or E -2-butene; II to Z ; and III to E .
(Section 7.5). Double-bond migration in propene may be followed by labelling either of the terminal carbon atoms with 13C. The reaction
CH3––CH |
|
CH2 → CH3––CH |
|
CH2 |
(7.D) |
|
|
||||
|
|
occurs rapidly on Au/SiO2 in the presence of hydrogen, and more slowly in Ag/SiO2;23 metals of Groups 8 and 10 have not yet been examined.
A besetting problem with the study of alkene hydrogenation is deactivation of the catalyst by strongly-adsorbed ‘carbonaceous deposits’2,24−26 or ‘acetylenic residues’11 that form even well below room temperature; the base metals of Groups 8 and 10 are especially prone to this, but even the noble metals are not immune, and even the most fundamental studies are necessarily made on ‘equilibrium’ surfaces, much of which is permanently inactivated.27−29 This raises the vexed question of whether such poisoned surfaces truly reflect the character of the metal, and indeed whether it is not on the carbonaceous overlayer that the catalysis occurs.30,31 Another idea that has been seriously suggested is that reaction actually occurs, in
HYDROGENATION OF ALKENES AND RELATED PROCESSES |
297 |
Figure 7.3. Temperature-dependence of equilibrium concentrations of the n-butene isomers.22
the case of supported metals, on the support, by spillover catalysis;32−34 the fact that unadulterated metals are also efficient catalysts does not automatically invalidate this hypothesis. These ideas will be considered in Sections 7.2.1 and 7.2.7.
7.2. HYDROGENATION OF ETHENE AND PROPENE4,6,15,35−37
It is convenient to treat these two molecules side-by-side because, they are the only ones with which addition and exchange are the only real possibilities. The preponderance of the literature concerns ethene, but where comparison can be made there is little to choose between the ways in which they behave, save for self-evident differences due to the numbers of carbon atoms.
7.2.1. Kinetics of Hydrogenation
The purpose of this section is to provide an overview of the principal kinetic features of the hydrogenation of ethene and of propene, as providing a framework (or at least part of one) within which discussion of mechanisms must be conducted. Their reactions with hydrogen (and with deuterium) are quite comparable; the addition of the methyl group leads to somewhat higher reactivity, due to weaker chemisorption as might be predicted from its lower heat of hydrogenation (Table 7.1). Relative rates for other alkenes will be considered later. The problem of deactivation by ‘carbon deposition’ has already been mentioned, but quantitative

298 |
CHAPTER 7 |
information is scarce, and experience differs: thus various platinum catalysts are reported to lose 25% of their initial activity in about 1 h, before stabilising,38 but in another case with similar catalysts there appeared to be no problem.39 Deactivation seems to be more serious with the base metals than with the noble metals, although removing oxygen from the ethene is beneficial,40 and when working with a constant volume system the order of adding the reactants is important; with nickel film, rates are slowest when ethene is introduced first and fastest and least reproducible when hydrogen is added first, intermediate values being found with simultaneous addition.41 This is easily understood in terms of the formation of ethylidyne or other decomposition products being inhibited by pre-adsorbed hydrogen atoms, the effect of which is only slowly removed.
It has been seriously suggested that the structure insensitivity of ethene hydrogenation42 and the constancy of the apparent activation energy finds an explanation if the reaction actually proceeds on top of a layer of carbonaceous species, which act as a bridge by which hydrogen atoms are carried from the metal to the ethene.43 This concept has however never been quantitatively modelled, nor does it account for the marked differences in behaviour between palladium and platinum that will appear shortly. A similar concept was adopted by Somorjai, who suggested44 that ethylidyne was the intermediate concerned (on Pt(111)); and although this species is now firmly relegated to the status of a mere spectator,45−47 its role and that of carbon in general to facilitate movement of hydrogen atoms has never been totally eliminated. It must therefore be remembered that, in the absence of information to the contrary, the metal will be at lease partly covered by strongly retained dehydrogenated species that may (or may not) play some role in the reaction.
By far the greater part of published work has been performed in flow systems, which is in some ways unfortunate because the dependence of rate upon time (i.e. real time as opposed to contact time) is not then accessible: the time course kinetics do not always correspond to the initial rate orders because of a slow response of surface concentrations to pressure changes, and mechanism may alter as the reactants are consumed.41 Orders of reaction are generally expressed as simple exponents, and only rarely (and usually only for ethene) is Langmuirian formalism used.2,48 Sometimes the former method only describes the results over a very limited pressure range.38 All of the most detailed experimental studies have been made with platinum catalysts of various descriptions, and from a broader viewpoint, considering also the reaction with deuterium, this is understandable, because ethene exchange is slight and its occurrence is indeed often ignored in otherwise comprehensive theoretical treatments.38 By comparison what we know of the reaction on palladium, nickel and other metals is much less detailed.
Alkene hydrogenation is considerably exothermic (Table 7.1), and maintaining isothermal conditions in a catalyst bed is, except at lowest conversions, not a

TABLE 7.2. Kinetic Parameters for the Hydrogenation of Ethene on Platinum Catalystsa
Form/Support |
Wt. % Pt |
E /kJ mol−1 |
PH /kPa |
PE /kPa |
x |
y |
T/K |
Notes |
References |
/SiO2 |
6.3 |
67 |
6.7 |
14.7 |
1 |
0 |
253 |
b |
39 |
/SiO2 |
1.2–11.5 |
29–50 |
97.5 |
2.5 |
— |
— |
— |
c |
55 |
/SiO2 |
0.04 |
34.5 |
20 |
3.3 |
0.5 |
−0.17 |
223 |
|
38 |
/SiO2 |
— |
35 |
— |
— |
0.77 |
−0.25 |
233 |
|
51 |
/SiO2 |
12 |
88 |
6.6 |
6.6 |
1.0 |
−0.2 |
333 |
d |
29 |
/SiO2 |
0.05 |
67 |
20 |
30 |
0.5 |
0 |
317 |
|
251 |
/Al2 O3 |
5 |
63 |
13 |
13 |
1.0 |
−0.35 |
423 |
e |
103 |
/Al2 O3 |
1 |
41 |
8 |
6.5 |
1.2 |
−0.5 |
273 |
f |
54 |
(111) |
— |
45 |
2.6 |
1.3 |
1.3 |
−0.6 |
315 |
g |
44 |
Foil |
— |
42 |
13 |
2.6 |
1.3 |
−0.8 |
323 |
|
2, 44 |
Wire |
— |
34.5 |
20 |
3.3 |
0.95 |
−0.33 |
336 |
|
38 |
Wire |
— |
42 |
13 |
2.6 |
1.2 |
−0.5 |
323 |
|
233 |
a See ref. 2 for pre-1960 results.
b Similar results were reported for 0.75% Pt /SiO2, and for Pt/Al2 O3 and Pt/MoO3.
c No systematic variation of E with either metal content or dispersion (see Figure 7.5). d Catalyst deliberately deactivated.
e Experiments conducted with D2 .
f Similar orders were reported for Pt foil and Pt on other ceramic supports.
PROCESSES RELATED AND ALKENES OF HYDROGENATION
299

300 CHAPTER 7
TABLE 7.3. Kinetic Parameters for the Hydrogenation of Ethene on Other Metals
Metal |
Form |
E /kJ mol−1 |
x |
y |
|
T/K |
References |
||
Fe |
/SiO2 |
35 |
0.91 |
−0.04 |
303 |
51 |
|||
Fe |
Film |
30.5 |
0.87 |
−0.6 |
305 |
41 |
|||
Co |
/SiO2 |
35 |
0.55 |
−0.19 |
213 |
51 |
|||
Ni |
/SiO |
2 |
35 |
0.67 |
−0 |
. |
08 |
233 |
51 |
|
|
a |
|||||||
Ni |
/SiO2 -Al2 O3 |
50 |
1.09 |
0.21 |
401 |
40 |
|||
Ni |
Film |
42 |
1→0.7 |
−0.4 |
305 |
41 |
|||
Cu |
/SiO2 |
35 |
0.69 |
0.06 |
353 |
51 |
|||
Ru |
/γ -Al2 O3 |
36 |
1 |
−0.2 |
327 |
108 |
|||
Ru |
/SiO2 |
35 |
0.95 |
−0.59 |
203 |
51 |
|||
Rh |
/SiO2 |
35 |
0.85 |
−0.74 |
197 |
51 |
|||
Pd |
/SiO2 |
35 |
0.66 |
−0.03 |
243 |
51 |
|||
Pd |
Al2 O3 |
— |
0.9 |
−0.3 |
298 |
47 |
|||
Os |
/γ -Al2 O3 |
35 |
1 |
— |
297 |
108 |
|||
Ir |
/Al2 O3 |
58 |
1.4 |
−0.6 |
333 |
103 |
|||
aE falls to 27 kJ mol−1 above 408 K, y increases to 0.36.
See 2 for pre-1960 results.
trivial matter. Ignoring it will vitiate kinetic measurements, and lead to unjustifiable conclusions.40
Notwithstanding the many differences of metal, dispersion, surface structure, cleanliness etc, there is a remarkable uniformity in the kinetic parameters observed in the region of room temperature. Of course even with a single metal this uniformity is not complete, and there are occasional cases of the rate (expressed per unit of active area) being badly out of line,49 perhaps due to unsuspected poisoning of the catalyst, and of activation energies being anomalously high50 or low.2 A selection of some of the more recent values (and a few older ones) is given in Tables 7.2 and 7.3, but in general those published before about 1960 are omitted; for these an earlier summary must be consulted.2 Turnover frequencies given for platinum in older work have been shown as an Arrhenius plot, giving a mean activation energy of 36 kJ mol−1. These provide a useful norm against which subsequent results can be compared; thus the TOF at 273 K should be49 about 1 s−1, but a tabulation of values for rates or TOFs is not particularly useful because of the variety of circumstances in which they have been measured, and the uncertainty of correcting them to any standard condition.
Activation energies are very frequently between 30 and 50 kJ mol−1, and for different metals in comparable physical form it is sometimes said that the same value suffices for all.12,51 Orders in hydrogen are always positive and very often are unity, while orders in ethene are usually either zero or sometimes negative. So general is this behaviour that attention is naturally drawn to the few exceptions. With platinum, orders of reaction are essentially independent of the physical form38 and the support,52 but are temperature-dependent (Table 7.4), the order in