

Metal-Catalysed Reactions of Hydrocarbons / 09-Hydrogenation of Alkynes
.pdf9
HYDROGENATION OF ALKYNES
PREFACE
The theme of the previous chapter is continued, because once again the reactions to be considered are dominated by the much stronger adsorption of the reactant compared to the products; high selectivities in the hydrogenation of alkynes to alkenes are frequently met. Emphasis is now placed rather on the causes of this high selectivity, rather than as before on the structure of the alkenes produced; the main product is often the one formed by simple addition of two hydrogen atoms to the same side of the alkyne. Because of the importance of selective alkyne hydrogenation in petrochemical operations and in organic synthesis, reaction mechanisms have been deeply researched, and much effort has been devoted to technical improvements to catalyst performance.
9.1. INTRODUCTION
9.1.1. The Scope of the Literature
The hydrogenation of alkynes has been widely studied, and there is a correspondingly large literature. Much of this concerns ethyne, and the vast majority of the papers deal with its reaction on palladium, which is outstanding for its high activity in this reaction, and for the generally high selectivity with which ethene can be formed from it. The motivation for this work has been the necessity in industrial practice to remove small amounts of alkynes (and dienes) from alkene streams produced by steam-cracking of hydrocarbons (see later). A subsidiary factor if the suitability of palladium for semi-hydrogenation of alkynes in the fine chemicals sector. Much of the work has been conducted in flow systems to mimic industrial practice, although the use of static systems has revealed puzzling aspects of the kinetics that still await full explanation.
395

396 |
CHAPTER 9 |
The emphasis in this Chapter will be on the factors that determine the degree of selectivity with which the intermediate alkene is formed; whereas in the previous Chapter, it lay more on the nature of the products formed, this is of lesser importance here, because under most circumstances the major product is that which arises from the addition of two hydrogen (or deuterium) atoms to the same side of the chemisorbed alkyne, i.e. Z-addition. Minor amounts of the products of E-addition do occur, and the mechanism by which they are formed merits discussion. However in the case of ethyne itself, this consideration is irrelevant unless deuterium is used, so the overwhelming thrust of the work on its hydrogenation has been towards understanding the mechanism by which ethene is so selectively formed. Further significant aspects of the reaction are best discussed after a brief description of the industrial problems.
9.1.2. Industrial Applications of Alkyne Hydrogenation1–6
The petrochemical industry is largely based on the conversion of alkenes containing mainly two to four carbon atoms into products of greater value by selective oxidation, polymerisation and other processes, for which purpose alkenes streams containing very low concentrations of alkynes and alkadienes (preferably <5 ppm) are required. Alkenes are produced by non-selective thermal processes (steam cracking) or catalytic cracking of naphtha fractions, and are separated into cuts containing predominantly molecules of a specified number of carbon atoms. However each cut contains the multiply unsaturated impurities (alkynes and dienes), so the problems of their selective removed by hydrogenation are similar. Much work has been done with ethyne, and a lesser but still significant amount with 1-butyne, where because of the lower volatility of the C4 fraction the reaction can be conducted in the liquid phase or in solution (Section 9.3.2).7 Two types of ethene feedstock are used, both containing 0.2 to 2% ethyne, but differing in their hydrogen and ethene contents: in the front-end cut the hydrogen content exceeds 15%, but in the tail-end cut it is only 0.5 to 3%. Typical reactant concentrations used in laboratory work designed to mimic industrial conditions are shown in Table 9.1,4 although these have been varied widely in explorations of their effects on performance.8−10
The technical success of a palladium catalyst is measured by its ability to lower the alkyne or diene content almost to zero without hydrogenating any of the alkene: indeed, ideally its content ought to rise slightly. Thus the alkene selectivity Se may take either positive or negative values, depending on whether it is being created or destroyed. Since the ethene: ethyne ratio may be as high as 345, this is a very tall order indeed for any catalyst, and in addition, with the C4 cut, 1-butene is the main component and the desired product, so that doublebond migration must not occur. Although palladium is the best metal it is not ideal for the following reasons. (1) Its selectivity is not perfect, and under some

HYDROGENATION OF ALKYNES |
397 |
laboratory conditions (e.g. high H2/C2H2 ratio) it can fall catastrophically. (2) The reaction is accompanied by the formation of oligomers, which foul up the plant (Section 9.3.3). (3) As we have seen in earlier chapters, it is among the most active metals for alkene isomerisation. Nevertheless its high activity and intrinsic selectivity make it the metal of choice, and in industrial practice is often used as 0.04% Pd/Al2O3; in laboratory work this concentration has been varied (0.005 to10%) to examine particle size and related effects. The selection of such a low concentration is, as we shall see, dictated not only by considerations of cost. Much research has been directed to overcoming these limitations, and two lines in particular have been followed: (i) the use of a selective poison added to the feedstock (carbon monoxide is most often used,11 Section 9.3.4); and (ii) the use of palladium-containing bimetallic catalysts (Section 9.3.5).
In the fine chemicals sector there is frequently the need to hydrogenate a molecule containing one or more C C bonds with high degrees of stereoand regioselectivity. Examples of this include the synthesis of insect sex pheromones,11 and of vitamin A. Here also the inherent high selectivity to partially reduced products shown by palladium is employed, but as before it is beneficially reinforced by selective poisons and appropriate choice of support (Section 9.3.5).
C bonds with high degrees of stereoand regioselectivity. Examples of this include the synthesis of insect sex pheromones,11 and of vitamin A. Here also the inherent high selectivity to partially reduced products shown by palladium is employed, but as before it is beneficially reinforced by selective poisons and appropriate choice of support (Section 9.3.5).
9.1.3. The Chemisorbed State of Alkynes
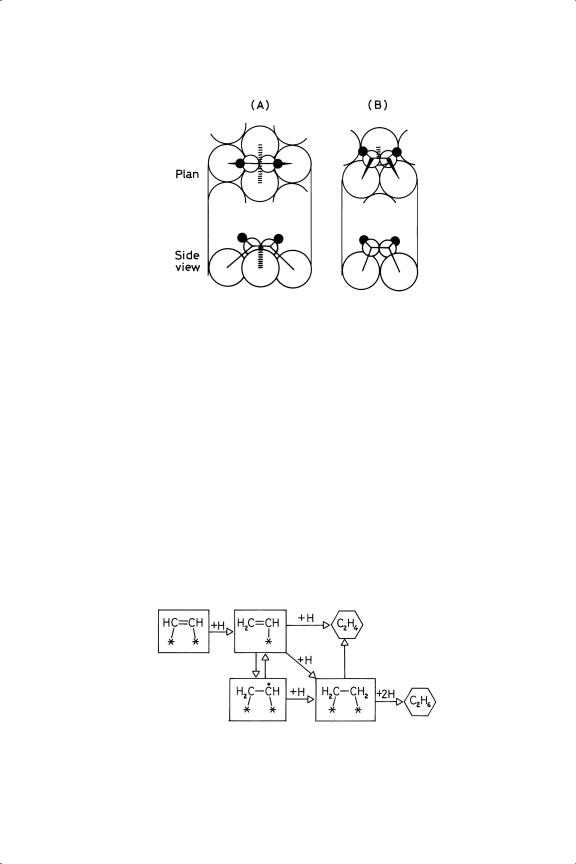
There have been a number of studies of the chemisorption of ethyne, employing FTIR, RAIRS, LEED, HREELS, STM and PED.13 The structures discovered have been reviewed in Section 4.4.3, so only a brief recapitulation is needed. These investigations reveal two basic structures, coded types A and B (respectively structure 15 and 13 in Table 4.2). In Type A, the H––C––C––H plane is parallel with the surface, and the molecule is held by two σ and two π bonds (di-σ /di-π ): on fcc(111) surfaces, the carbon atoms lie above two adjacent trigonal holes, and so the molecule forms a ‘long bridge’ as shown in Figure 9.1. In Type B, the plane is tilted as a consequence of only a single π bond at one side (di-σ /π ) (Figure 9.1); this should represent a slightly weaker mode of bonding. There is little evident chemical logic in which surfaces show which structure. The low index faces of copper give only Type A, as does the (100) face of all three Group 10 metals: Ni(110) and Pd(110) show Type B, as do the (111) faces of palladium and platinum; Ni(111) gives Type A.13 There is clearly scope for a thorough theoretical study of the bonding of ethyne to metal surfaces. It has to be remembered however that this work is usually carried out at low temperature, and so does not necessarily tell us the forms that are reactive in hydrogenation at ambient temperature and above. A RAIRS study of propyne on Ni(111) and Cu(110) suggests structures analogous to those shown by ethyne:14 on Ni(111) the C––C bond order in a Type A structure has been lowered to almost unity, and the structure is stable from 110 to 293 K.
398 |
CHAPTER 9 |
Figure 9.1. Structures of ethyne chemisorbed on fcc(111) surface: (A) long bridge; (B) short bridge.105
9.1.4. The Origin of Selectivity in Alkyne Hydrogenation
Work described in the literature can be divided into two broad categories:
(i)the hydrogenation of an alkyne by itself (including the use of deuterium as an isotopic label, Section 9.2.4), and (ii) its hydrogenation in the presence of a large excess of the product alkene, in order to simulate industrial practice (including
the use of compounds labelled with radioactive carbon). There are some intermediate cases, where smaller amounts of the alkene have been added.15−17 Work of the second type ventures into areas that are met in the first type only at very high conversions, and are therefore usually ignored: important and difficult questions are thereby raised that are irrelevant to the hydrogenation of the alkyne alone.
In this latter category, it was long ago suggested that two factors are relevant: these are illustrated by the simplified reaction scheme shown as Scheme 9.1. Less than complete selectivity to ethene occurs if either (i) chemisorbed ethene fails to desorb, and remains on the surface long enough to be hydrogenated to ethane, or
(ii)ethene that has desorbed can re-adsorb in competition with the ethyne. These
Scheme 9.1. Simplified scheme for the mechanism of ethyne hydrogenation. Note: in this and the following Schemes no attempt is made to show all interactions of π orbitals with the surface.

HYDROGENATION OF ALKYNES |
399 |
two factors are readily distinguished by observing how the selectivity alters with conversion of the ethyne: if it remains constant, then the first explanation applies, whereas if it continually decreases,18 the second explanation has to be invoked. These two possibilities were termed respectively mechanistic and thermodynamic factors.16 Of course, as the Scheme shows, both may operate at the same time, but the thermodynamic factor will assume greater importance towards the end of the reaction, when the ethene/ethyne ratio has become large, or when additional ethene has been deliberately added.
Under industrial conditions the mechanistic selectivity is not of prime importance, because the ethyne concentration is so low; but it is disastrous if the ethene can adsorb in competition with the ethyne, and become hydrogenated to ethane. The amount of ethane arising from the two sources can easily be distinguished by labelling the ethyne with either 13C or 14C. What is therefore needed is a very large thermodynamic factor, i.e. for the ethyne to be much more strongly adsorbed than the ethene. If this situation is secured, not only will access of ethene to the surface be prevented by low concentrations of ethyne, but the ethyne will displace the chemisorbed ethene formed by its hydrogenation. The two selectivity factors are therefore closely linked. Thus if selectivity to ethene in the hydrogenation of ethyne alone is high and independent of conversion until almost the end of the reaction, it is likely that excess ethene will not gain access to the surface. On the other hand,18 when the selectivity is not very high even at the outset, as sometimes happens with rhodium and iridium, it falls as conversion increases, because the ethene formed is able to compete successfully for a share of the surface. This is not always the case, however.15
It will also become clear that the behaviour of alkynes closely resembles that of the 1,2- and 1,3-alkadienes already discussed. Their strong chemisorption was due to the simultaneous interaction of both double bonds in the latter case, and probably to the large release of strain that accompanied the opening of one of the double-bonds (or at least the disengagement of one set of π orbitals) in the latter case. The heat of hydrogenation reflects the magnitude of the release of strain and hence the strength of the interaction with surface atoms: for ethyne to ethene it is 172 kJ mol−1 compared to 137 kJ mol−1 for ethene to ethane. These reactions are thus quite exothermic, and in industrial use care must be taken to avoid temperature excursions, as these would lead to loss of selectivity.
9.1.5. Interpretation of Results: Some Preliminary Comments
As the results are presented in more detail in the following sections, it will be natural to wonder what further development of the mechanistic framework in Scheme 9.1 will be needed to account for them. Such considerations are postponed to Section 9.3.2, but the following points should be borne in mind. The reaction system palladium-ethyne-hydrogen is in fact extremely complex,19 and there are

400 |
CHAPTER 9 |
many complicating aspects barring the way to simple explanations. It is simplistic to try to assign a particular observation to a single cause, as it is more probable that a number of factors are simultaneously at work. In particular it is notoriously difficult to isolate a single parameter of the system (e.g. metal particle size) and to study this by itself, because other things change at the same time. Taking this variable as an example, we have seen (Section 2.5) that consequential geometric and electronic effects cannot meaningfully be separated, although this is sometimes attempted. More importantly, the tendency to form the β-hydride phase is size-sensitive,19 and dissolved hydrogen atoms have been implicated in the reaction;19−21 the likelihood of deactivation by carbonaceous deposits22−25 or by formation of a carbide phase26 may also change, and the former have been assigned important roles in determining selectivity. As particle size is lowered, spillover becomes more probable, as does a metal-support interaction. Interpretation of results is therefore a minefield through which we must walk delicately.
9.2. HYDROGENATION OF ETHYNE: 1, IN STATIC SYSTEMS27–29
9.2.1. Introduction
The classic work on this reaction, published between about 1945 and 1970,16−18,30−37 was mainly conducted in constant-volume reactors, the progress of the reaction being followed by the pressure fall. Forced recirculation of the reactants through the catalyst bed was occasionally used, but more usually trust was placed on natural convection, which was usually adequate as rates were comparatively slow. Only rarely was the effect of added ethene noted.16−17 This modus operandi had the advantage that the effect on product yields of a smoothly varying conversion was immediately apparent, and certain interesting and informative kinetic phenomena were observed that were inaccessible to flow systems.30−32 Its disadvantage is that the rate and kinetic form of the reaction depends somewhat on the initial conditions such as the order in which the reactants are introduced. If the hydrocarbon is added first, the surface may become partially coated with stronglyadsorbed derived species before the hydrogen is introduced; if the reverse sequence is used, some time may elapse before surface concentrations attain their steady state. Work of this type is the concern of this section, which also covers studies made in UHV systems.
Much of the work designed to elucidate or to improve upon palladium catalysts in the industrial context has naturally been performed in continuous-flow systems, sometimes employing a spinning basket;38 the effect of added ethene has been a recurrent theme. In such work the catalyst is usually allowed to reach its steady level of activity before results are taken, or conditions altered to ascertain their effects. Work of this kind is summarised in Section 9.3.
HYDROGENATION OF ALKYNES |
401 |
9.2.2. Kinetic Parameters
In the study of non-catalysed reactions, there are several methods available for determining orders of reaction, and when properly performed they lead to the same results. It is therefore disconcerting to find that this is not always the case with heterogeneously catalysed reactions, the hydrogenation of ethyne being a case in point. In a closed system, the variation of rate with extent of reaction ought to be governed by the remaining pressures of the reactants raised to the power of the order, viz.
r = k PH x PC y |
(9.1) |
the values of x and y being determined from the way in which the initial rate depends on the pressure of each reactant varied separately. If one reactant is in large excess, its pressure will not change much as the reaction proceeds, and so the rate is chiefly dependent upon the pressure of the other reactant. These concepts are fully explained in all textbooks of physical chemistry.
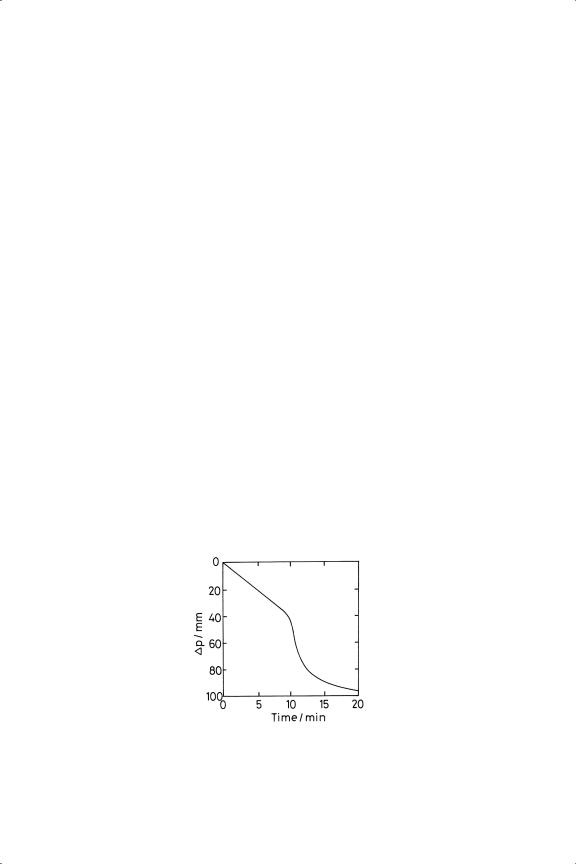
It has been observed routinely that when ethyne is admitted first to a vessel containing a supported metal of Group 10, and hydrogen is then added such that the hydrogen: ethyne ratio ( PH/PC) is two or more, the rate remains constant until at some point it starts to accelerate (Figure 9.2), notwithstanding the initial rate method giving the orders in hydrogen and ethyne as respectively about one and zero (see Table 9.1).16,17,27,28,30−34,39 The acceleration, which is most marked with nickel and palladium, starts when the ethene/ethyne ratio has risen to the point where ethene can compete with the ethyne for space on the surface and can itself by hydrogenated; and, because its rate of hydrogenation is much faster than that of
Figure 9.2. Hydrogenation of ethyne over Pd/pumice at 293 K in a constant volume system; pressure fall versus time ( PE = 50 Torr; PH =150 Torr).33

402 CHAPTER 9
TABLE 9.1. Kinetic Parameters for the Hydrogenation of Ethyne in the Absence of Added Ethene: r PHx PE y
Metal |
Form |
E /kJ mol−1 |
x |
y |
T/K |
S2,i |
S0 |
References |
Fe |
/pumice |
64 |
1 |
0 |
429 |
91 |
30 |
28,36 |
Co |
/pumice |
17 |
1 |
0 |
470 |
90 |
50 |
28,36 |
Ni |
Powder |
— |
1 |
−0.5 |
403 |
— |
— |
28 |
Ni |
/pumice |
51 |
1 |
0 |
353 |
83 |
60 |
28,36 |
Ni |
/pumice |
43 |
1 |
−0.14 |
371 |
85 |
— |
30 |
Cu |
/pumice |
38 |
1 |
0.3 |
473 |
90 |
60 |
28,36 |
Cu |
/various |
88 |
1 |
−0.1 |
450 |
99 |
— |
171 |
Pd |
/pumice |
50 |
1 |
−0.5 |
322 |
92 |
25 |
28,36 |
Pd |
/A12 O3 |
46 |
1 |
−0.5 |
273 |
97 |
— |
17 |
Pd |
/α-A12 O3 |
— |
1.42 |
— |
293 |
96 |
— |
16 |
Pd |
/SiO2 |
71 |
1 |
0 |
387 |
97 |
— |
39 |
Pd |
/SiO2 |
— |
— |
— |
293 |
94 |
— |
15 |
Pd |
Foil |
40 |
1.04 |
— |
300 |
31 |
— |
43 |
Ag |
/SiO2,TiO2 |
39 |
— |
— |
353 |
100 |
0 |
66 |
Au |
Al2 O3 |
34 |
0.4 |
0.1 |
360 |
100 |
30 |
67 |
Pt |
/pumice |
50 |
1.2 |
−0.7 |
346 |
82 |
|
28,36 |
Pt |
/Al2 O3 |
39 |
1.5 |
−0.7 |
383 |
90 |
28 |
34 |
Rh |
/pumice |
65 |
1 |
0 |
358 |
86 |
25 |
37 |
Rh |
/A12 O3 |
38 |
1.4 |
0 |
403 |
90 |
— |
18 |
Rh |
/SiO2 |
— |
— |
— |
293 |
74 |
— |
15 |
Ir |
/A12 O3 |
— |
1 |
−0.3 |
403 |
55 |
15 |
34 |
Ir |
/SiO2 |
— |
— |
— |
293 |
16 |
|
15 |
Ru |
/Al2 O3 |
44 |
1 |
0 |
385 |
90 |
8 |
41 |
Os |
/Al2 O3 |
33 |
1 |
0 |
398 |
65 |
16 |
41 |
E is the activation energy for total reaction: values for the formation of C2 products and of oligomers may differ slightly. T/K is the temperature at which the orders and initial C2 selectivities (S2,i ) were measured : S0 is a rounded value for the oligomer selectivity, which in most cases was obtained at about T/K . Further details of earlier work (<1960) are to be found in references 27 and 39. See also 16 and 85.
ethyne, acceleration occurs.16,40 The acceleration point therefore depends on the selectivity to ethene in the early part of the reaction, and for this reason it occurs sooner as either PH or temperature is raised.17,27,34 With platinum, however, the acceleration is more gradual, and occurs at least initially without loss of selectivity; it is due to the negative order in ethyne (Table 9.1). Zero-order pressure-time curves have been found in butadiene hydrogenation over the Group 10 metals,31 and with ethyne hydrogenation over copper,32 but not over the metals of Groups 8 and 9:18,32,41 with these metals the rate decreases as the reaction proceeds, in line with the initial rate law, although acceleration can still sometimes be seen.41 When the initial PH/ PC ratio is less than two, the rate decreases continuously, being determined by the instantaneous pressures of the reactants;40 no acceleration is observed if there is insufficient hydrogen left to hydrogenate the ethene. Careful and detailed studies on the base metals of Groups 8 to 10,32 especially nickel,30,31
HYDROGENATION OF ALKYNES |
403 |
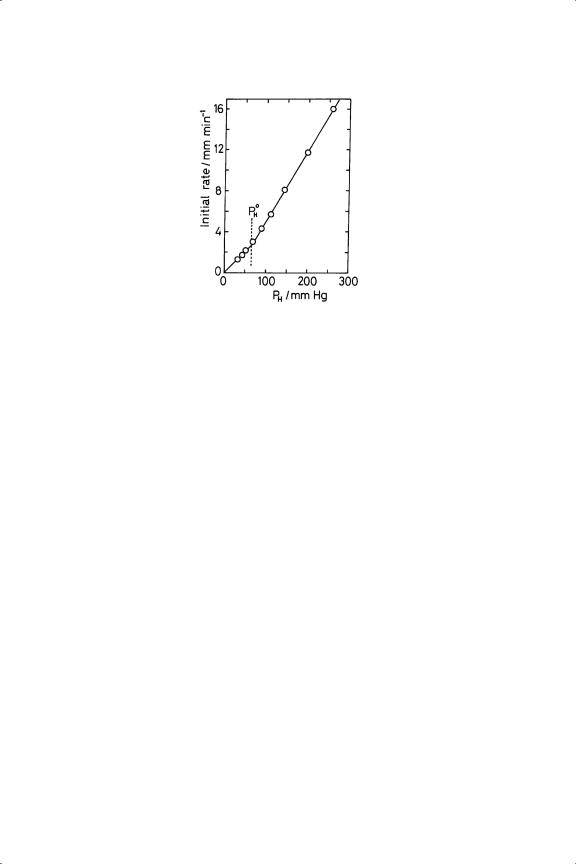
Figure 9.3. Hydrogenation of ethyne over Ni/pumice at 486 K in a constant volume system: dependence of initial rate on PH ( PE = 30.5 Torr).30
have revealed sudden slight changes of rate during the zero-order phase when hydrogen was admitted first.
The insensitivity of the rate to the varying hydrogen pressure after prior admission of ethyne was (and is) most puzzling, but there are further surprises to come. Although rates were proportional to hydrogen pressure when PH/PC ratio was less than two, in the case of the base metals at higher pressures the zeroorder rate constant assumed a greater value (Figure 9.3), so the full rate expression becomes
r = k A PH + kB ( PH − PH0) |
(9.2) |
where PH0 is about twice PC.30,31 Variation of temperature showed that the corresponding activation energies E A and E B on Ni/pumice were respectively 43 and 100 kJ mol−1. Even more surprising was the observation that the sudden injection of more hydrogen or ethyne during the zero-order phase caused the rate to change in accordance with the initial rate law for the latter and equation 9.2 for the former.30 These strange findings have received no further attention since they were first disclosed. They were discussed in some detail at the time,16,30−32 but no firm conclusion was reached: it seems most likely that when a sufficiently high pressure of hydrogen is present a certain concentration of reactive centres is set up, and this is sustained by some kind of chain reaction while reactant pressures decrease slowly. Fragmentary results suggest that the same effect may be seen with other
404 |
CHAPTER 9 |
metals,42 but no systematic studies have been reported. A further comment will be offered later when the mechanism is re-considered, but the mystery remains.
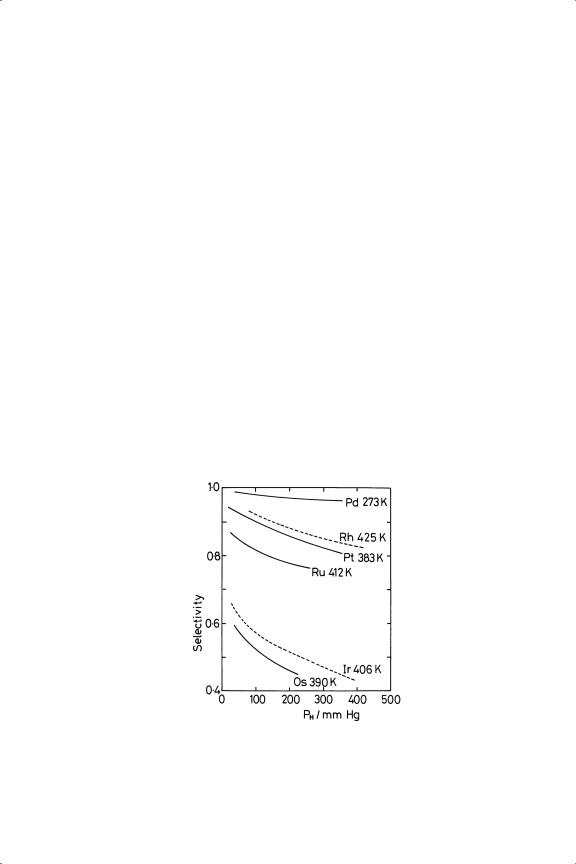
The principal kinetic features shown by the metals of Groups 8 to 10 and copper have marked similarities, which can be summarised as follows: (1) Orders in hydrogen are usually close to first, although with platinum34 and rhodium,18 and palladium (especially at higher temperatures),16,17,43 they are significantly greater, approaching 1.5 (see Table 9.1). (2) Orders in ethyne are most often close to zero, but especially with palladium and platinum they can be substantially negative (see also Table 9.1), suggesting some degree of competition between the reactants for the surface. (3) Activation energies are mainly between 30 and 50 kJ mol−1, with only few exceptions (Table 9.1): this is the range in which true values can be expected, so the composition of the reactive layer cannot be very temperaturedependent. (4) Ethene selectivities vary considerably, but are high (>80%) in most cases, especially with palladium (except in the form of foil43), but are markedly lower with osmium and iridium15,44,45 (Table 9.1): they are however somewhat lower than those shown by butadiene27,46 (Table 8.6). (5) They decrease with increasing hydrogen pressure (Figure 9.4) and decreasing temperature27 (Figure 9.5), as expected intuitively, since higher concentrations of hydrogen in the reactive layer must favour formation of ethane. (6) All metals catalyse the formation of oligomers; their yields vary very much with reaction conditions; the activation energy for their formation is higher than that for C2 products, and the process is inhibited by increasing hydrogen pressure. Over the base metals of Groups 8 to
Figure 9.4. Hydrogenation of ethyne over Al2 O3 -supported metals: effect of PH on selectivity ( PE = 50 Torr).27