

Квантово-химические расчеты органических молекул - ustu012
.pdfФедеральное агентство по образованию ГОУ ВПО «Уральский государственный технический университет – УПИ»
В.А. Бакулев, Ю.Ю. Моржерин, Ю.О. Субботина
КВАНТОВО-ХИМИЧЕСКИЕ РАСЧЕТЫ ОРГАНИЧЕСКИХ МОЛЕКУЛ
Учебное электронное текстовое издание Научный редактор проф., д-р хим. наук В.С. Мокрушин
Методическое указание по проведение квантово-химических расчетов в пакете программ CHEMOFFICE/CHEM3D для студентов дневной формы обучения специальностей 655500 – Биотехнология, 070100 – Технология биоорганического синтеза, 655000 Химическая технология органических веществ, 250101 Химическая технология основного органического синтеза, 250103 Химическая технология органических красителей.
Предназначены для студентов 3 и 4 курса специализации 655500 - Биотехнология, 070100 – Технология биоорганического синтеза, 655000 Химическая технология органических веществ, 250101 – Химическая технология основного органического синтеза, 250103 – Химическая технология органических красителей при прохождении ими учебно-исследоватеского практикума по курсам «Основы научного исследования», «Компьютерное моделирование органических молекул». Методические указания включают программы коллоквиумов и методики практических занятий.
© ГОУ ВПО УГТУ−УПИ 2004
Екатеринбург
2005
СОДЕРЖАНИЕ |
|
Практикум.................................................................................................................... |
3 |
Практическая работа №1. «Основные навыки работы с программой |
|
CHEMOFFICE/CHEM3D»....................................................................................... |
3 |
Практическая работа №2. «Оптимизация геометрии. Алгоритмы |
|
оптимизации» ......................................................................................................... |
17 |
Практическая работа №3. «Оценка точности расчетов зарядового |
|
распределения на основании расчетов дипольного момента».......................... |
19 |
Практическая работа № 4. «Определение центров нуклеофильной и |
|
электрофильной атаки в ряду моно- и ди-замещенных бензолов» .................. |
20 |
Практическая работа №5. «Изучение механизма электроциклических |
|
процессов на примере 1,3-бутадиена. Конротаторный и дисротаторный тип |
|
электроциклизации» .............................................................................................. |
22 |
Практическая работа №6. «Изучение стабильности различых состояний |
|
аминокислот в газовой фазе»................................................................................ |
25 |
Литература рекомендуемая для подготовки к практическим занятиям............. |
27 |
2
ПРАКТИКУМ
Практическая работа №1
«Основные навыки работы с программой CHEMOFFICE/CHEM3D»
Цель работы. Научится использовать интерфейс программы
Вопросы к допуску.
1.Как создать новую папку на жестком диске с заданным именем?
2.Как сохранить файл в заданной папке? Как переименовать файл? Как скопировать файл? Как удалить файл?
3.Как открыть файл (папку)?
Ход работы.
I.Основные принципы работы с программой Chem3D
Как открыть рабочее окно программы? Из меню Пуск выбрать закладку Про-
граммы, а затем выбрать папку ChemOffice. В ней выбрать закладку Chem3D. Графический интерфейс программы (Рисунок 1) очень удобен позволяет сделать расчеты наглядными для пользователя. Интерфейс состоит из окна для изображения модели, строки меню и команд, набора различных инструментов.
Окно модели служит рабочим пространством, где проводится моделирование молекул. Это пространство включает в себя, кроме окна для отображения модели, строку состояния и панель вращения. Текстовая информация о модели выводится в окне для вывода сообщений. Ниже, в таблице приводятся типы задач, для которых используются элементы окна модели.
3
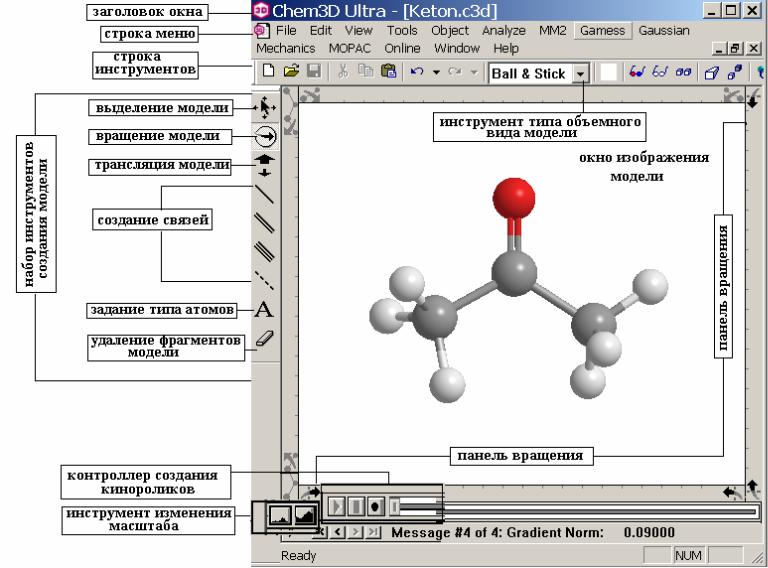
Рисунок 1. Графический интерфейс программы Chem3D.
4

|
Элемент |
Пояснение |
|
|
|
|
|
Рабочее пространство, где выводится изображение модели, |
|
|
проводится процесс ее построения и преобразования. Точка от- |
|
Окно модели |
счета в системе декартовых координат (0,0,0) помещается в |
|
центре окна, в не зависимости от того, как развернута модель и |
|
|
|
каков ее масштаб. Оси декартовых координат остаются непод- |
|
|
вижными по отношению к окну модели. |
|
|
|
|
Строка |
Отображает информацию об активных границах вашей модели |
|
|
|
|
состояния |
и о скрытых атомах модели. |
|
|
|
|
Панель |
Производит вращение вокруг различных осей в зависимости от |
|
|
|
|
вращения |
того, какой атом был выделен. |
|
|
|
|
Заголовок |
Отображает имя файла, под которым сохранена модель. |
|
окна |
|
|
Строка меню |
Содержит меню операций, которые можно произвести над мо- |
|
делью. |
|
|
|
|
Панель масштабирования позволяет изменять размеры модели.
|
Элемент панели |
|
|
Пояснение |
|
|
масштабирования |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Уменьшение |
|
Уменьшает масштаб. Щелкнуть один раз для уменьше- |
||
|
|
ния на 10%. |
|||
|
|
|
|
||
|
|
|
|
|
|
Увеличение |
Увеличивает масштаб. Щелкнуть один раз для увели- |
|
чения на 10%. |
||
|
Набор инструментов для создания модели позволяет создавать модели и управлять ими.
5
II.Построение модели
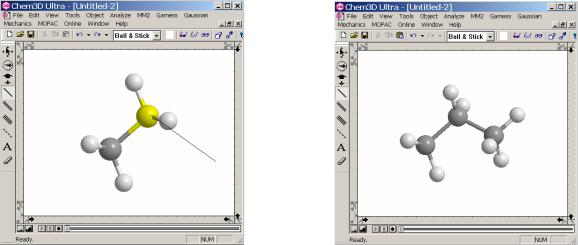
Для создания новой модели необходимо из меню Файл (File) выбрать Создать (New model). Затем приступить непосредственно к построению модели. В качестве примера здесь приводится процедура построения молекулы ацетона.
1. Создание углеродного остова модели.
На первом шаге создается углеродный остов модели. Для того чтобы создать связь необходимо использовать инструмент создания связей. При использовании данного инструмента создаются только углерод - углеродные связи.
Для этого надо щелкнуть по инструменту одинарной связи  , нарисовать связь в окне, вытянув ее в желаемом направлении (щелчок по полю окна и удерживая левую клавишу мыши протянуть и отпустить клавишу). Для того чтобы добавить связи к модели необходимо выбрать из панели инструмент связей. На поле модели щелкнуть по атому, к которому следует добавить связь. И, удерживая левую клавишу мыши протянуть ее в нужную точку. После чего отпустить клавишу. При стандартных настройках при появлении атомов углерода автоматически будут добавлены водородные атомы, в соответствии со степенью гибридизации углеродных атомов.
, нарисовать связь в окне, вытянув ее в желаемом направлении (щелчок по полю окна и удерживая левую клавишу мыши протянуть и отпустить клавишу). Для того чтобы добавить связи к модели необходимо выбрать из панели инструмент связей. На поле модели щелкнуть по атому, к которому следует добавить связь. И, удерживая левую клавишу мыши протянуть ее в нужную точку. После чего отпустить клавишу. При стандартных настройках при появлении атомов углерода автоматически будут добавлены водородные атомы, в соответствии со степенью гибридизации углеродных атомов.
Шаг 1. |
Шаг 2. |
6
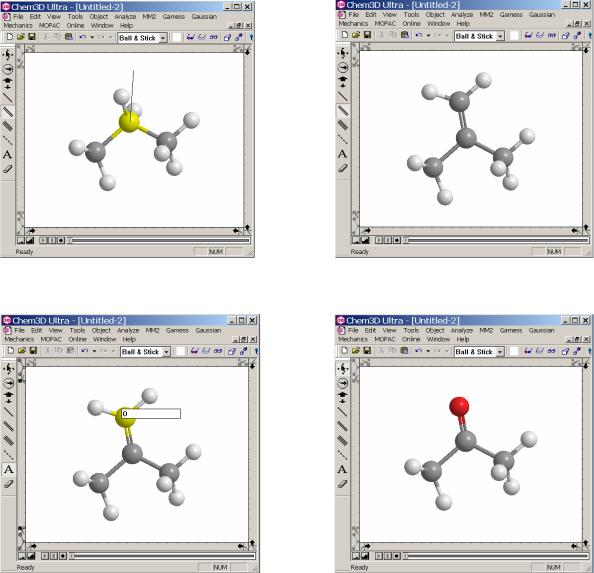
2. Введение гетероатомов в модель
После того как углеродный остов молекулы создан можно, добавляя гетероатомы, преобразовать модель. Чтобы ввести гетероатом следует использовать следующий прием: первоначально создать дополнительную связь инструментом создания связей (шаг 3,4). Затем, выбрать из набора инструментов инструмент задания типа атомов и щелкнуть по атому, в положение которого вводится гетероатом (шаг 5). После чего появится ячейка с мигающим курсором для ввода текста. В этой ячейке необходимо набрать символ гетероатома, после чего нажать на клавиатуре клавишу Enter. В результате атом углерода (водорода) будет заменен гетероатомом (шаг 6).
Шаг 3. |
Шаг 4. |
Шаг 5. |
Шаг 6. |
7
3. Дополнительные функции
Убрать лишние элементы можно при помощи кнопки  , выбрав ее на панели инструментов и щелкнув по ненужному фрагменту модели.
, выбрав ее на панели инструментов и щелкнув по ненужному фрагменту модели.
Для выделения фрагментов модели используют инструмент выделения  .
.
Чтобы выставить заряд в молекуле (например, для цвиттерионов) можно использовать два следующих приема:
-Используя инструмент текста. Щелкнуть на атом, на котором предполагается расположение заряда, затем набрать на клавиатуре «+» или «-». Набранный заряд автоматически фиксируется программой для дальнейших расчетов.
-Используя ключевые слова. Из меню MOPAC выбрать Минимизация энергии (Minimize Energy). Затем, в меню Общее (General ), в ячейке Допол-
нительные ключевые слова (Additional Keywords) набрать ключевое слово
CHARGE=n, где n отвечает положительному или отрицательному заряду (-2, - 1, +1, +2).
При этом стоит отметить, что зарядовое распределение, полученное двумя разными методами в некоторых случаях будет различаться.
III. Квантово-химический расчет. MOPAC
Программа MOPAC предназначена для проведения полуэмпирических расчетов различных молекул. Программа имеет ограничение по применению, так как содержит необходимые параметры только для 250 тяжелых атомов. Описываемые ниже процедуры расчета предполагают наличие знания основной терминологии (оптимизация молекулы, минимизация энергии, точечные расчеты).
Восновном выделяют следующие типы задач для расчетов:
-минимизация энергии
8
-поиск переходных состояний
-расчет различных свойств молекул (дипольный момент, тензоры, зарядовое распределение и прочее)
1.Поиск минимума энергии модели, расчет свойств молекул.
Запуск программы Минимизация энергии это самая первая и необходимая процедура, которую тре-
буется выполнить. Это является залогом успеха получения корректных данных по другим свойствам молекулы. Поэтому, прежде чем переходить к расчету свойств молекул, необходимо минимизировать энергию модели.
Чтобы минимизировать энергию необходимо из меню MOPAC, выбрать Минимизация энергии (Minimize Energy) (шаг 7). После этого появляется диалоговое окно процедуры. Диалоговое окно содержит четыре подменю: тип задачи (Job type), тип теории (Theory), свойства(Properties), общее (General).
В подменю тип задачи (Job type) (шаг 8) можно установить набор опций, контролирующих вывод на экран результатов расчета, а также задание минимального значения градиента. Какие еще опции следует выбирать в подменю тип задачи (Job type)?:
Если вы хотите... |
Тогда... |
|
|
Задать критерий точности |
Следует набрать на клавиатуре Минимальное зна- |
поиска минимума на по- |
чение градиента (Minimum RMS Gradient). Если |
верхности потенциальной |
вы хотите задать величину <0.01, тогда также не- |
энергии |
обходимо задать LET в секции дополнительных |
|
ключевых слов. Если «горка» на ППЭ становится |
|
очень незначительной, то процедура минимизации |
|
останавливается. В этом случае говорят о дости- |
|
жении локального минимума. По умолчанию, ве- |
|
личина градиента равная 0.100 представляет собой |
|
рациональный выбор, как в пользу точности, так и |
|
в пользу времени расчета. Уменьшение этой вели- |
|
чины приводит к замедлению расчетов, так как чем |
|
ближе к области минимума, тем больше времени |
|
требуется для достижения его в этом случае. Так |
|
же увеличение этой величины не приводит к |
|
улучшению в плане точности расчетов. Наоборот, |
|
чем больше градиент, тем дальше от положения |
|
|
|
9 |
|
минимума. Однако, в случае конформационного |
|
|
анализа это может оказать неоценимую помощь. |
|
|
Конформации молекулы обладают стабильностью, |
|
|
но различаются по энергии. Поэтому, выставив |
|
|
очень маленький градиент, в результате миними- |
|
|
зации будет найдена конформация отвечающая са- |
|
|
мой низкой энергии. |
|
|
|
|
Наблюдать процесс ми- |
Отображение каждого шага (Display Every Itera- |
|
нимизации «вживую» на |
tion). NВ: Отображение или сохранение данных |
|
каждом шаге оптимиза- |
каждого о шага минимизации приводит к сущест- |
|
ции |
венному увеличению времени расчетов. |
|
|
|
|
Записать каждый шаг ми- |
|
|
нимизации как кадр ви- |
Запись результатов каждого шага (Record Every |
|
деоизображения для |
Iteration). |
|
повторного воспроизве- |
||
|
||
дения. |
|
|
|
|
|
Отображать результаты |
Копировать измерения в виде сообщения (Copy |
|
расчетов в виде сообще- |
||
ния. |
Measurements to Messages) . |
|
|
||
|
|
Подменю тип теории (Theory) (шаг 9) используется для задания метода и типа используемой волновой функции. Для этого необходимо, во-первых, выбрать подходящий метод расчетов (АМ1, РМ3 и т.д.), а, во-вторых, подходящую волновую функцию. Выбор функции из Wave Function меню заключается в вы-
боре между ограниченным (RHF) и неограниченным (UHF) методом Хартри-
Фока (Closed Shells и Open Shells). По умолчанию, для систем с закрытой и открытой оболочек выставлен ограниченный метод Хартри-Фока. Если же модель имеет открытую электронную оболочку, то в ограниченном методе Хартри-Фока будет задействовано приближение полуэлектрона и метод КВ
(конфигурационное взаимодействие) для синглетных микросостояний системы. Неограниченный метод Хартри-Фока является альтернативой и может быть применен для расчета молекул, как с закрытой оболочкой, так и открытой, но требует как минимум в два раза больше времени для расчета.
Чтобы задействовать ограниченный метод Хартри–Фока следует выбрать волновую функцию Закрытая оболочка (Closed Shell (Restricted)). Чтобы приме-
10