

-
Изоляторы: кислотные и основные катализаторы.
Катализаторы, относящиеся к этой группе, не проводят электрический ток (по крайней мере при относительно умеренных температурах катализа), то есть не имеют свободно перемещающихся электронов, и поэтому практически не катализируют редокс-реакции.
Например, твердые оксиды элементов 3-го периода (Na2O, MgO, Al2O3, SiO2, P2O5) являются изоляторами. Как видно, в этом ряду слева направо свойства оксидов меняются от основных через амфотерные до кислотных. Подобным образом ведут себя оксиды других периодов.
Возникающие при хемосорбции заряды на поверхности катализаторов-изоляторов не передаются по катализатору, а локализованы на активных центрах. Поэтому каталитические свойства изоляторов не могут быть в достаточной степени объяснены электронной теорией, а наиболее подходящей концепцией для объяснения каталитических свойств изоляторов является концепция кислот и оснований.
Хорошо известными и наиболее используемыми в промышленном органическом синтезе кислотно-основными катализаторами являются: Al2O3, алюмосиликаты, цеолиты, MgO, SiO2/MgO, силикагели, алюмофосфаты и специальные глины, активированные химической обработкой.
Все эти катализаторы содержат на поверхности кислотные центры.
Доля основных промышленных гетерогенных катализаторов гораздо меньше, чем доля кислотных. В Таблице 5.21 приведены известные кислотно-основные катализаторы.
Таблица 5.21.
Классификация кислотно-основных катализаторов.
|
Гетерогенные кислотные катализаторы |
Гетерогенные основные катализаторы |
|
|
-
Кислотные катализаторы.
Оксид алюминия.
Оксидные катализаторы с кислотной поверхностью катализируют многие промышленно-важные реакции (дегидратация спиртов, гидратация олефинов, крекинг, полимеризация олефинов, конденсация карбонильных соединений, этерификация и пр.).
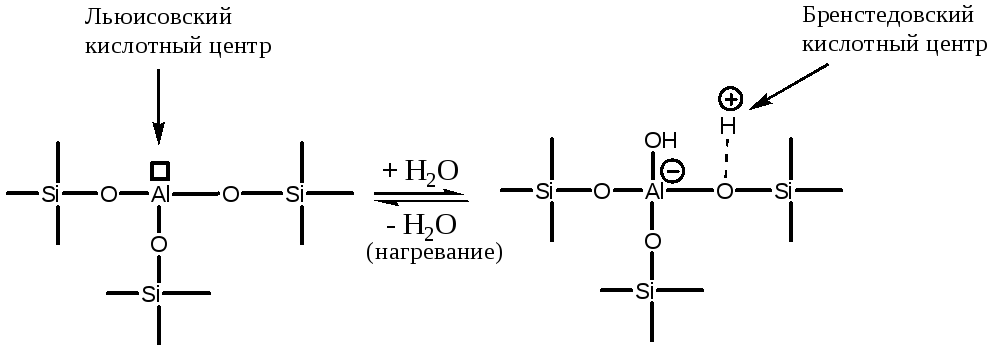
Кислотные центры на поверхности твердых оксидов могут обладать как протонной кислотностью (Бренстеда), так и апротонной кислотностью (Льюиса) (См. п. 2.1.1).
Рассмотрим природу протонных и апротонных кислотных центров на примере оксида алюминия. Al2O3 всегда содержит связанную воду, количество которой сильно зависит от температуры. Свежеприготовленный (свежеосажденный) Al2O3 полностью гидроксилирован на поверхности вплоть до температуры 100оС. В таком состоянии поверхностные -ОН группы ведут себя как слабые кислоты Бренстеда:
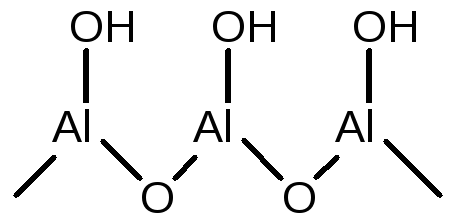 (5.56)
(5.56)
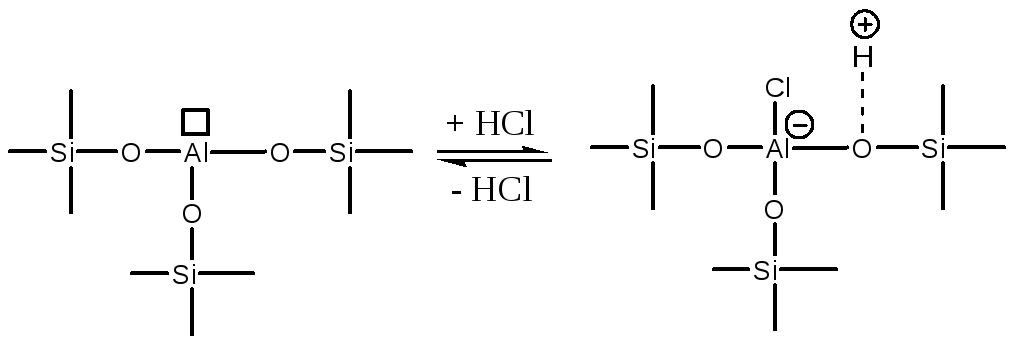
Значительно увеличить кислотность Бренстедовских центров можно замещением части гидроксильных групп на хлорид или фторид анион (обработкой соответствующим галогеноводородом):
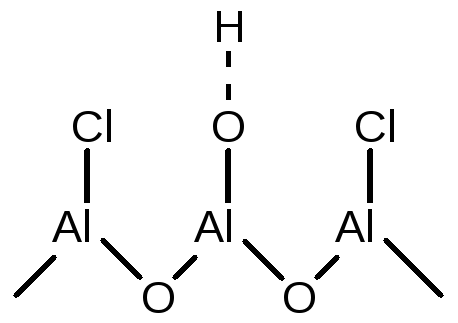 (5.57)
(5.57)
Увеличение кислотности -ОН группы объясняется сильным индукционным эффектом галоген-внионов, находящихся рядом с -ОН группой.
При нагревании гидроксилированного оксида алюминия выше температуры 150оС происходит дегидратация поверхностных и приповерхностных -ОН групп. При этом образуются дегидроксилированные атомы алюминия на поверхности, обладающие средней Льюисовской кислотностью:
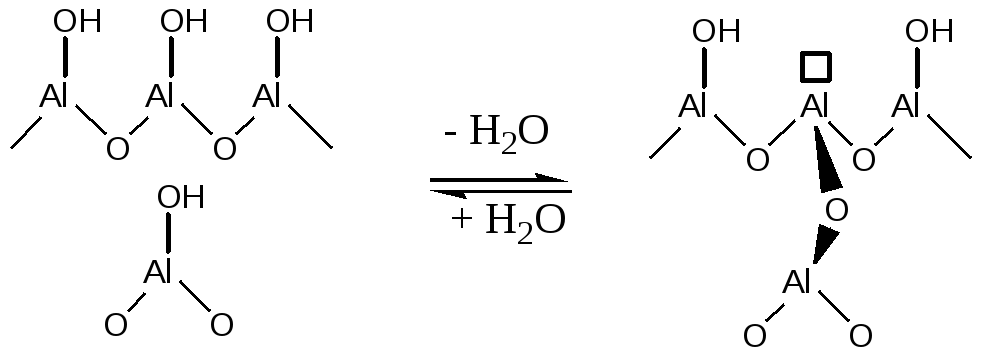 (5.58)
(5.58)
Одновременно с образованием таких Льюисовских центров усиливается кислотность соседних Бренстедовских центров за счет возможного донорно-акцепторного взаимодействия следующего типа:
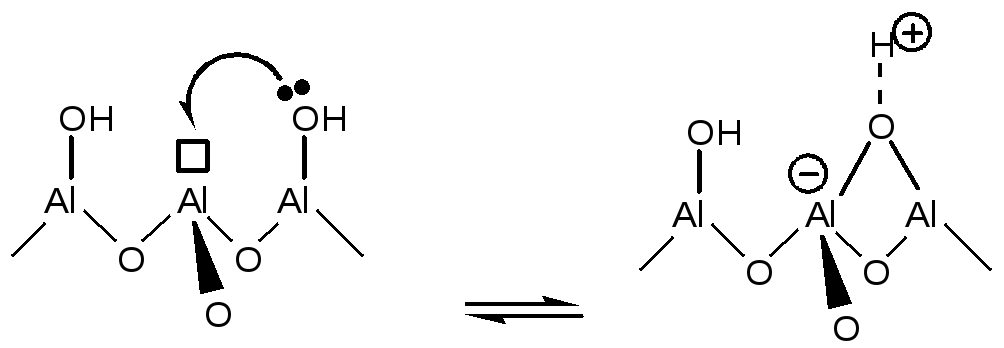 (5.59)
(5.59)
При температуре 400оС и выше происходит дегидроксилирование поверхностных атомов алюминия с образованием сильных Льюисовских кислотных и основных центров:
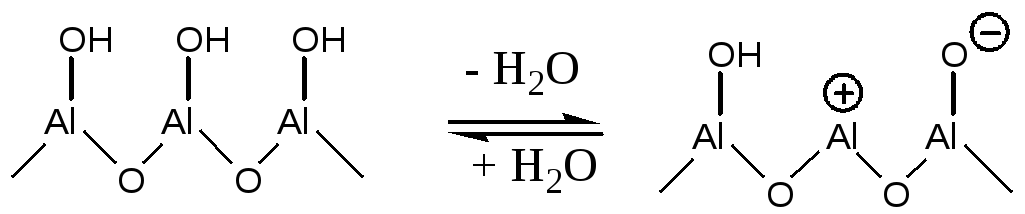 (5.60)
(5.60)
При температуре 900оС поверхность Al2O3 дегидроксилируется полностью и содержит только Льюисовские кислотные и основные центры.
Регулирование силы и соотношения протонных и апротонных кислотных центров, а также основных центров на поверхности Al2O3 очень важно для обеспечения оптимального каталитического эффекта в той или иной реакции. Установлено, что сильные Бренстедовские кислотные центры играют определяющую роль для реакций полимеризации олефинов, диспропорционирования толуола до бензола и ксилолов. Льюисовские кислотные центры являются определяющими в реакции деструкции алканов.
Циклизация олефинов в процессе реформинга протекает при согласованном катализе соседними - основным и протонным кислотным - центрами на поверхности оксида алюминия:
(5.61)
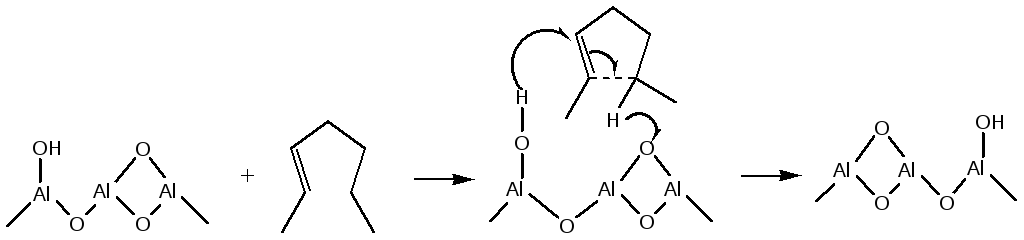

Каталитическое действие гетерогенных кислот и оснований такое же как и у гомогенных. Каталитическая активность коррелирует с кислотностью активных центров в соответствии с уравнением Бренстеда:
lgk = lgG - H0 (5.62)
Значения функции кислотности центров на поверхности гетерогенного катализатора можно получить экспериментально:
-
либо с помощью индикаторов Гаммета;
-
либо по ИК-спектрам адсорбированных оснований, таких как аммиак, пиридин, хинолин.
Второй способ основан на том, что комплексы выбранных оснований с кислотами Бренстеда и Льюиса имеют различное строение и поэтому имеют характерные полосы поглощения в разных областях спектра. Общее количество кислотных центров определяют по количеству хемосорбированного аммиака. Затем по интенсивности сигналов в ИК-спектрах можно определить соотношение концентраций протонных и апротонных кислотных центров на поверхности, а по смещению сигнала - силу соответствующих центров.
Например поверхностные соединения и соответствующие характерные частоты в ИК-спектре при использовании в качестве "зонда" пиридина следующие:
|
|
Бренстедовские центры (5.63) Ион пиридиния 1540 см-1 |
|
|
Льюисовские центры (5.64) кислотно-основный комплекс 1465 см-1 |
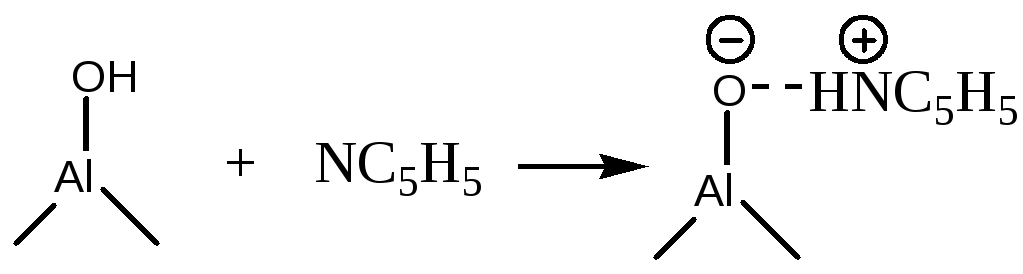
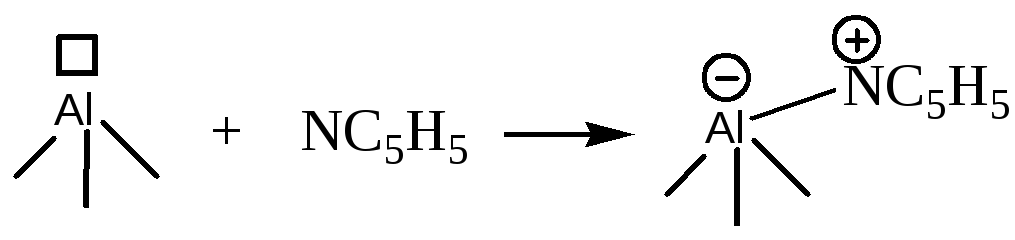
Оксид кремния.
Двуокись кремния (силикагель) является аморфным продуктом поликонденсации кремниевой кислоты с участием силанольных групп (Si-OH) и образованием силоксановых групп (Si-O-Si):
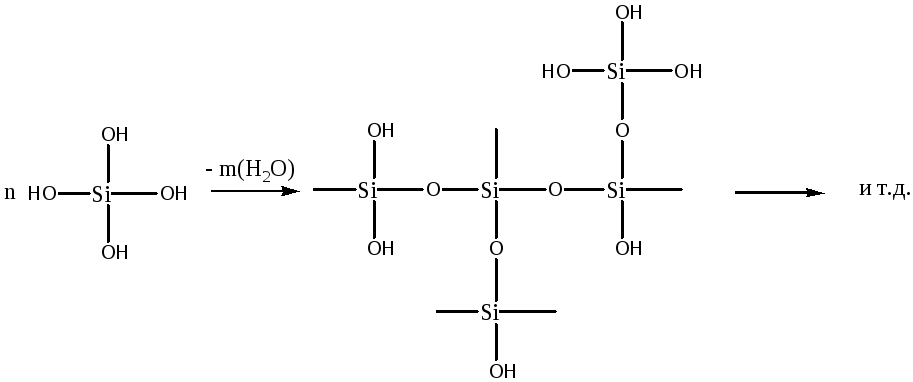 (5.63)
(5.63)
Связь гидроксильных групп с поверхностными атомами кремния в SiO2 гораздо сильнее, чем связь гидроксильных групп с атомами алюминия в Al2O3. Поэтому дегидратация силикагеля при температурах до 120оС сопровождается потерей только физически адсорбированной воды. Дегидроксилирование поверхности оксида кремния начинается только при температурах выше 200оС. При температуре 500оС концентрация поверхностных силанольных групп составляет около 20-30% от начальной, а при 1000оС - 10-15%.
Таким образом, в отличии от оксида алюминия, на поверхности SiO2 Льюисовские центры образуются не так охотно, и при температурах до 300оС практически не обнаруживаются. Поэтому на поверхности оксида кремния доминируют Бренстедовские центры, но кислотность их низка, и сравнима с кислотностью уксусной кислоты. Поэтому сам силикагель, как катализатор не представляет интерес, но является ценным носителем, так как обладает высокой удельной поверхностью (до 800 м2/г).
Алюмосиликаты.
Алюмосиликаты - один из важнейших типов гетерогенных кислотных катализаторов, применяемых в процессах крекинга.
По химическому строению они представляют собой двуокись кремния, в которой часть атомов кремния замещена на атомы алюминия. Трехзарядный ион алюминия на поверхности окружен тремя четырехзарядными ионами кремния, обладающими большей электроотрицательностью. Это приводит в сильному понижению электронной плотности на атоме алюминия и, следовательно, к увеличению его электрофильности (Льюисовской кислотности). В результате молекулы воды подвергаются на поверхности алюмосиликата диссоциативной хемосорбции:
 (5.64)
(5.64)
что приводит к образованию сильного протонного кислотного центра. При нагревании до высокой температуры происходит дегидратация поверхности и образование сильного Льюисовского кислотного центра (обратная реакция (5.64)).
Сильная кислотность алюмосиликатов обеспечивается уже при концентрации Al2O3 до 10-12%. Экспериментально установлено, что максимальная кислотность наблюдается при 20-30%-ном содержании Al2O3.
Увеличение кислотности алюмосиликатов можно достичь обработкой их небольшим количеством HCl:
 (5.65)
(5.65)
Соотношение концентраций кислотных центров Бренстеда и Льюиса на поверхности алюмосиликатов и их распределение по силе регулируют соотношением SiO2/Al2O3, температурой процесса, обработкой HCl и Na2O. Таким образом удается тонко регулировать активность и селективность катализируемых процессов.
Цеолиты.
Цеолиты, в отличии от Al2O3, SiO2 и алюмосиликатов, которые являются аморфными веществами, - представляют собой водосодержащие кристаллические вещества с высокоупорядоченной структурой. Основным строительным блоком цеолитов является тетраэдр из четырех анионов кислорода, окружающих ион Si4+ или Al3+ (Рис. 5.49). Заряд каждого иона кремния сбалансирован зарядами окружающих ионов кислорода, и, следовательно, кремний содержащие тетраэдры - электронейтральны. А алюминий содержащий тетраэдр имеет избыточный заряд -1, так как катион алюминия имеет заряд +3. Поэтому в состав цеолитов входят катионы, компенсирующие эти отрицательные заряды. Тетраэдры объединяются в блоки по связям -Si-О-Si-, из которых строится структура цеолита. В общем виде химический состав элементарной ячейки цеолита отображается формулой:
Mj/n[(AlO2)j (SiO2)y (H2O)z] (5.66)
где: М - катионы, как правило щелочных и щелочноземельных металлов; j - количество катионов алюминия в ячейке; n - заряд катиона М (таким образом j/n - количество катионов М, необходимое для компенсации отрицательных зарядов алюминийсодержащих тетраэдров); Y - количество ионов кремния; Z - количество гидратационной воды.
Упрощенно структура поверхности Na-содержащего цеолита представлена на Рисунке 5.50-а. Если натрий заменить на протон, то получится цеолит в Н+-форме (Рис. 5.50-б.).

Рис. 5.49. Кремний- и алюминий-содержащие тетраэдры в структуре цеолитов.
а 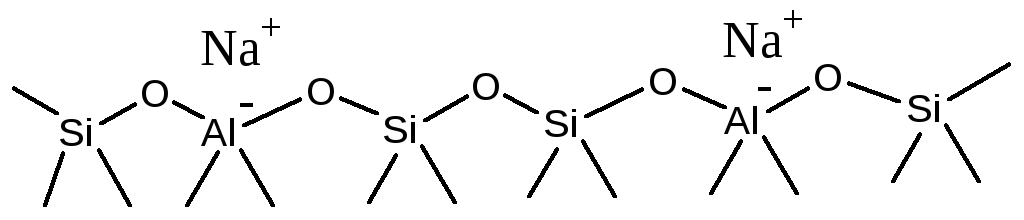
б 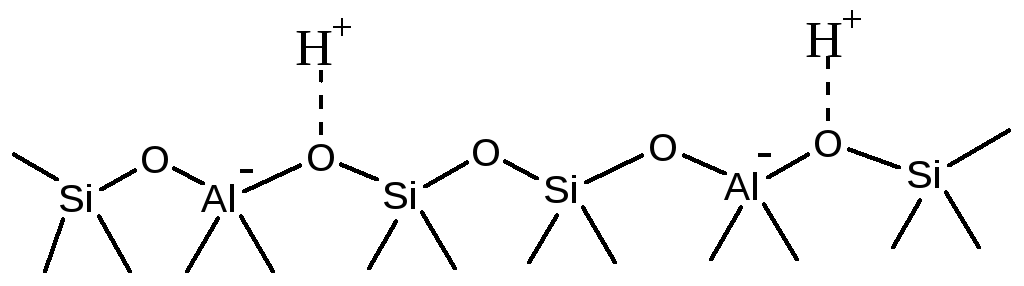
Рис. 5.50. Поверхность цеолита: а - в Na-форме; б - в Н-форме.
Регулируя при синтезе цеолитов соотношение Si/Al, природу катионов М, степень замещения М на Н+ и глубину удаления гидратационной воды кислотность цеолитом можно менять в очень широких пределах. Поэтому цеолиты являются ценными и широко применяемыми катализаторами кислотного типа в промышленной органической химии.
Благодаря уникальным свойствам применение цеолитов в катализе этим не ограничивается. Более детально цеолиты будут рассмотрены отдельно в п. ***.
Механизмы реакций, протекающих на поверхности кислотных катализаторов.
Внутримолекулярная и межмолекулярная дегидратация.
Кислотные катализаторы ускоряют как внутри-, так и межмолекулярную дегидратацию гидроксилсодержащих углеводородов. Активность и селективность катализаторов (помимо условий проведения процесса) зависит от кислотности поверхности.
Рассмотрим пример каталитических превращений этанола на различных образцах промышленного Al2O3, различающихся содержанием технических примесей - SiO2 и Na2O (Табл. 5.22).
Таблица 5.22.
Поведение оксида алюминия в дегидратации этанола.
|
Относительная кислотность* (при 175оС) |
Содержание примесей, %
|
Конверсия, % |
Селективность, % |
Кокс, % |
||
|
SiO2 |
Na2O |
С2Н4 |
(С2Н5)О |
|||
|
0,021 0,046 0,060 |
0,02 0,01 0,13 |
0,25 0,06 0,03 |
66,1 98,8 85,1 |
25,3 99,2 89,2 |
70,1 0,2 0,1 |
0,1 0,2 0,5 |
* - ммоль NH3, адсорбируемого 1 граммом Al2O3.
Проанализируем данные Таблицы 5.22. Увеличение содержания SiO2 приводит к усилению Бренстедовской кислотности. Увеличение содержания Na2O приводит нейтрализации сильных Бренстедовских кислотных центров, и к стабилизации основных центров. Очевидно, что общая кислотность образцов растет с увеличением содержания SiO2 и уменьшением содержания Na2O.
Установлено, что дегидратация этанола протекает при согласованном действии кислотных протонных и основных каталитических центров:
 (5.66)
(5.66)
Поэтому наивысшая активность и селективность по этилену наблюдается на образце со средней кислотностью (оптимальное соотношение основных и кислотных центров). Образец с наименьшей кислотностью обладает самой низкой активностью (конверсия) и наименьшей селективностью по этилену. Кроме того было установлено, что дегидратация протекает на кислотных центрах Бренстеда средней силы, а Льюисовские центры практически не катализируют эту реакцию. Этот вывод был сделан на основании того факта, что добавка небольших количеств оснований Льюиса (аммиак, пиридин и т.п.), блокирующих в первую очередь сильные кислотные и апротонные центры, не ингибируют реакцию.
Образование эфира на поверхности Al2O3 протекает по механизму Лэнгмюра-Хиншельвуда, при котором взаимодействие происходит между алкоголятом (С2Н5О-), образовавшемся на основном центре и молекулой этанола, активированной водородной связью на соседнем кислотном центре. Поэтому, чем больше содержание Na2O, тем выше селективность образования эфира.
Важный момент в данном примере - образование кокса. Как видно количество кокса растет пропорционально росту Бренстедовской кислотности. Это связано с тем, что именно на этих центрах образуются промежуточные карбокатионы, инициирующие димеризацию и олигомеризацию этилена. Отложение нелетучих продуктов олигомеризации приводит к блокированию поверхности, снижению каталитической активности и коксообразованию.
Реакции в каталитическом крекинге и реформинге (крекинг, изомеризация, полимеризация, циклизация, ароматизация, коксообразование)
Ключевой стадией, обеспечивающей каталитическую активность кислотных катализаторов в реакциях крекинга, изомеризация, полимеризация, циклизация, ароматизация, коксообразования, является образование карбокатионов при взаимодействии углеводородов с активными центрами поверхности.
Образование карбокатиона может протекать по трем механизмам:
-
Отрыв гидрид-аниона от парафина сильными кислотными центрами Льюиса:
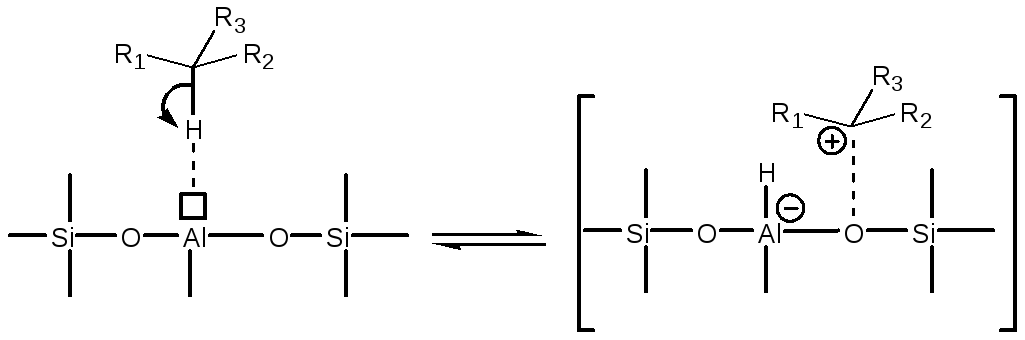 (5.67)
(5.67)
-
Протонирование парафина кислотными центрами Бренстеда с образованием водорода и карбокатиона. Такая реакция возможна только при участии очень сильных Бренстедовских центров, например, расположенных рядом с Льюисовским центром ((5.59), (5.64)):
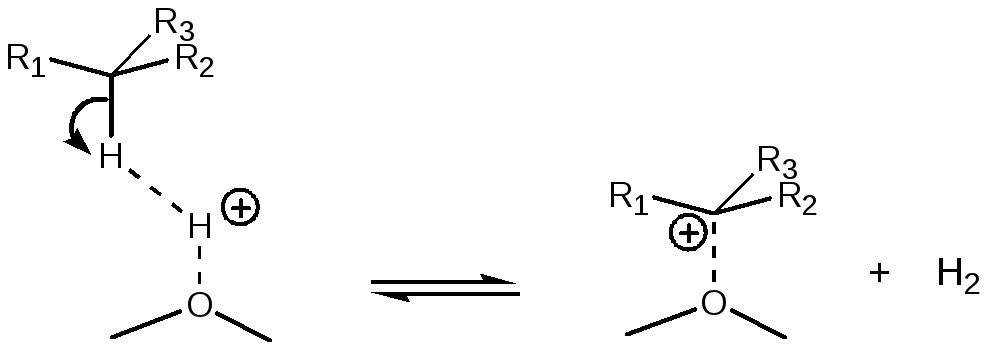 (5.68)
(5.68)
-
Протонирование олефина:
Н+ + R1-CH=CH-R2 R1-CH2-+CH-R2 (5.69)
Олефины, необходимые для протекания реакции (5.69) могут присутствовать в сырье как примесь, образовывать в результате термического крекинга или в результате крекинга карбокатионов, образующихся на каталитических центрах по реакциям (5.67), (5.68). Крекинг неразветвленного вторичного карбокатиона протекает по схеме:
R1-CH2-CH2-+CH-CH2-R2 R1-CH=CH2 + +CH2-CH-R2 (5.70)
Образовавшийся первичный карбокатион может перегруппироваться в более устойчивый вторичный или третичный.
Один из возможных механизмов циклизации и ароматизации углеводородов на кислотных катализаторах может быть представлен следующей схемой. Олефины способны отдавать аллильный гидрид-анион кислотным центрам катализатора с образованием резонансно-стабилизированного аллильного крбокатиона:
(5.71)
![]()
Образовавшийся карбокатион может отдавать протон основному центру каталитической поверхности с образованием диена:
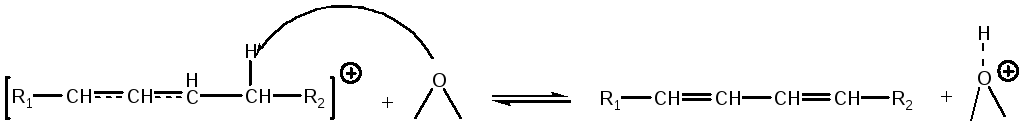 (5.72)
(5.72)
Далее по такому же механизму может образоваться триен, который легко циклизуется при катализе протонными кислотными центрами:
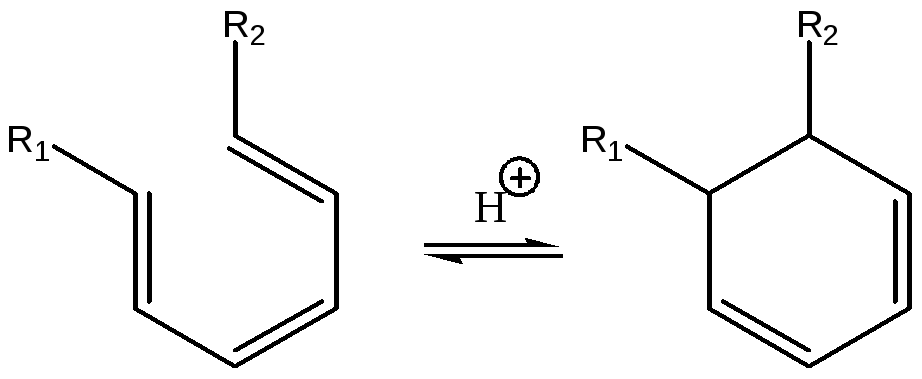 (5.73)
(5.73)
Образовавшийся циклический диен при превращается в карбокатион при взаимодействии с другим карбокатионом на поверхности катализатора:
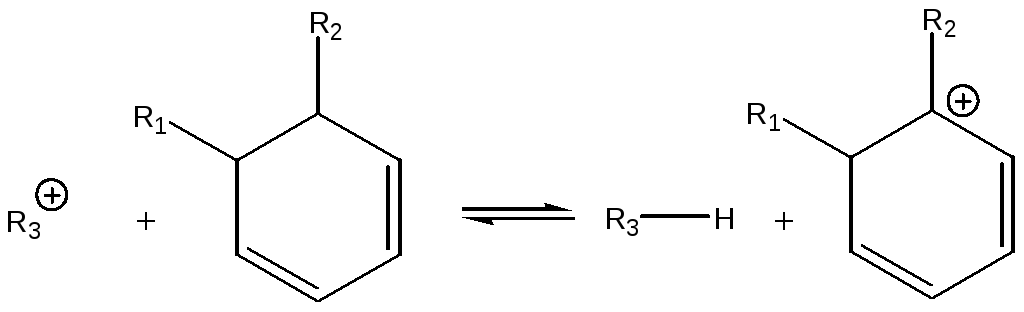 (5.74)
(5.74)
Далее, отдавая протон основному центру катализатора, крбокатион превращается в ароматический углеводород:
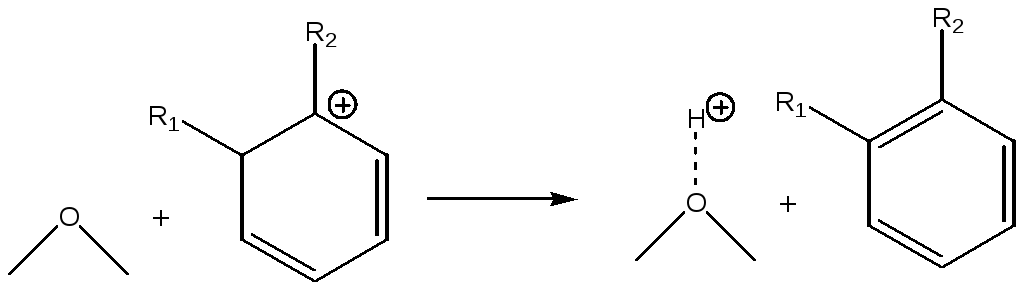 (5.75)
(5.75)
Циклизация может протекать и в результате внутримолекулярной электрофильной атаки в карбокатионе, имеющем двойную связь:
 (5.76)
(5.76)
Как видно из приведенных схем ароматизация углеводородов на кислотных катализаторах протекает с участием основных центров. Но так активность основных центров в кислотных катализаторах низка, то реакции ароматизации протекают медленно. При каталитическом крекинге газойля доля ароматических углеводородов в продуктах составляет порядка 2%.
Кислотно-каталитические реакции изомеризации, полимеризации, алкилирования, переалкилирования (диспропорционирования) на гетерогенных катализаторах протекают по тем же механизмам, что и при катализе гомогенными кислотами.
Образование кокса на поверхности кислотных катализаторов связано главным образом с кислотно-каталитической реакцией конденсации ароматических соединений. Упрощенная схема может быть представлена так:
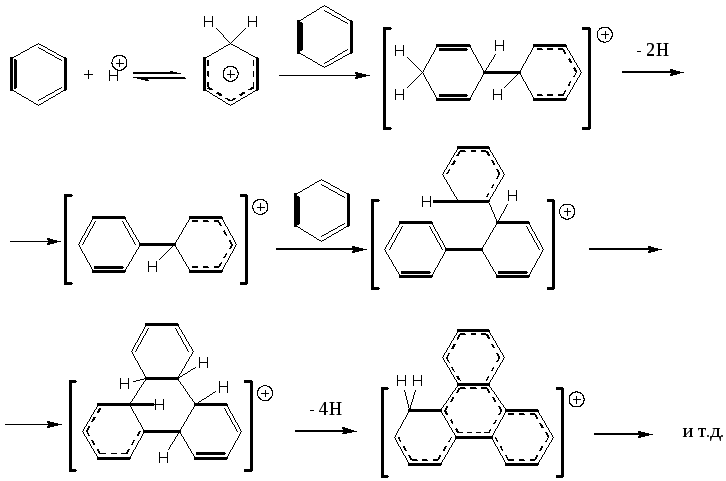 (5.77)
(5.77)
Многоядерный ароматический карбокатион очень устойчив, поэтому он может не только отдавать протон с образованием полиядерного соединения:
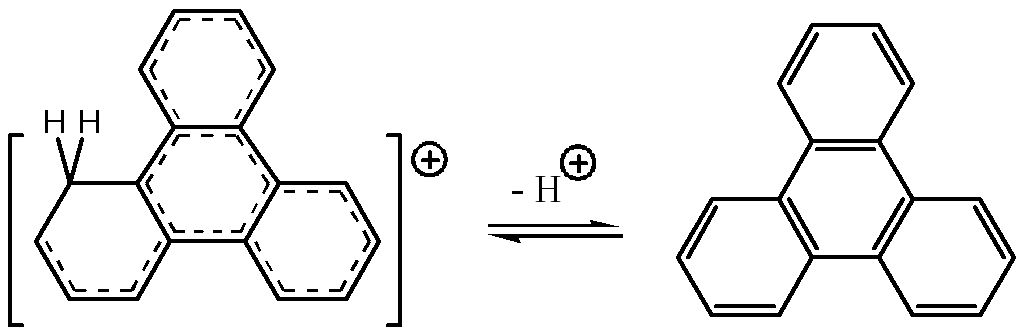 (5.78)
(5.78)
но и продолжать цепочку реакций конденсации дальше с образованием кокса. Эта упрощенная схема образования кокса может сопровождаться реакциями алкилирования, полимеризации, циклизации.
Кислотность катализаторов и реакционная способность.
В реакциях протекающих только на кислотных центрах (например, полимеризация олефинов) активность коррелирует с кислотностью поверхности (в соответствии с уравнением Бренстеда).
Опыт показывает, что наибольшая кислотность достигается на смешанных оксидах, в которых катионы имеют разное координационное число (или степень окисления). В Таблице 5.23 приведены смешанные оксиды с широким спектром кислотности поверхности. Каталитическая активность гетерогенных кислотных катализаторов соответствует каталитической активности гомогенных минеральных кислот с тем же значением кислотности. Но при этом гетерогенные катализаторы имеют преимущество по термической стабильности и легкости отделения от реакционной массы.
Таблица 5.23.
Сила кислотных центров двойных оксидов.
|
Состав компонентов А-В |
Содержание компонента А, % |
Удельная поверхность, м2/г |
Н0 |
|
Al2O3 - SiO2 ZrO2 - SiO2 Ga2O3 - SiO2 BeO - SiO2 MgO - SiO2 Y2O3 - SiO2 La2O3 - SiO2 |
94 88 92.5 85 70 92.5 92.5 |
270 448 90 110 450 118 80 |
- 8.2 ( 90% H2SO4) - 8.2 -7.2 - 8.2 -7.2 - 6.4 - 6.4 - 5.6 ( 71% H2SO4) - 5.6 - 3.2 |
В таблице 5.24 приведены интересные результаты, демонстрирующие влияние кислотности на показатели эффективности катализаторов в реакциях изомеризации полимеризации и крекинга.
Таблица 5.24.
Поведение кислотных катализаторов в различных реакциях в порядке увеличения кислотности.
|
Катализатор |
Изомеризация н-пентана (+ нанесенная Pt); Температура реакции, оС |
Полимеризация пропилена при 200оС; Конверсия, % |
Крекинг н-гептана; Температура достижения 10%-ной конверсии, оС |
|
-Al2O3 SiO2 ZrO2 TiO2 -Al2O3, низкая удельная поверхность -Al2O3, высокая удельная поверхность -Al2O3, хлорированный MgO - SiO2 Гетерополикислота -Al2O3, фторированный Алюмосиликат Цеолит (Н-форма) Н3РО4 на носителе AlCl3, HCl/Al2O3 |
не активен не активен не активен не активен
500
450
430 400
не стабильна 380
360 260 - 120 |
0 0 0 0
< 1
0-5
10-20 20-30
70-80 > 80
> 90 > 95 90-95 100 |
не активен не активен не активен не активен
не активен
490
475 460
не стабильна 420
410 350 не стабильна 100 |
Данные Таблицы 5.24 показывают, что каталитическая активность испытанных твердых кислот в выбранных трех модельных кислотно-каталитических реакциях меняется симбатно и увеличивается с увеличением кислотности.