

Обмен аминокислот 3
.pdfВыделяют 2 формы фенилкетонурии:
1) Классическая фенилкетонурия:
Причина: наследственный дефект фермента фенилаланингидроксилазы.
Частота заболевания: 1 случай на ~ 10000 новорожденных.
2) Вариантная фенилкетонурия: (коферментзависимая гиперфенилаланинемия)
Причина: мутации в генах, контролирующих метаболизм H4-биоптерина.
Встречается: 1-2 случая на ~ 1 млн. новорожденных.
H4-биоптерин необходим для гидроксилирования не только Фен, но и Тир и Три, поэтому, при этой форме заболевания нарушен метаболизм всех 3 аминокислот, а также синтез многих нейромедиаторов.
При этой форме заболевания возникают тяжелые неврологические нарушения и ранняя смерть.
Лечение фенилкетонурии: диета, с почти полным исключением из пищи фенилаланина.
! Начинать: сразу после рождения ребенка.
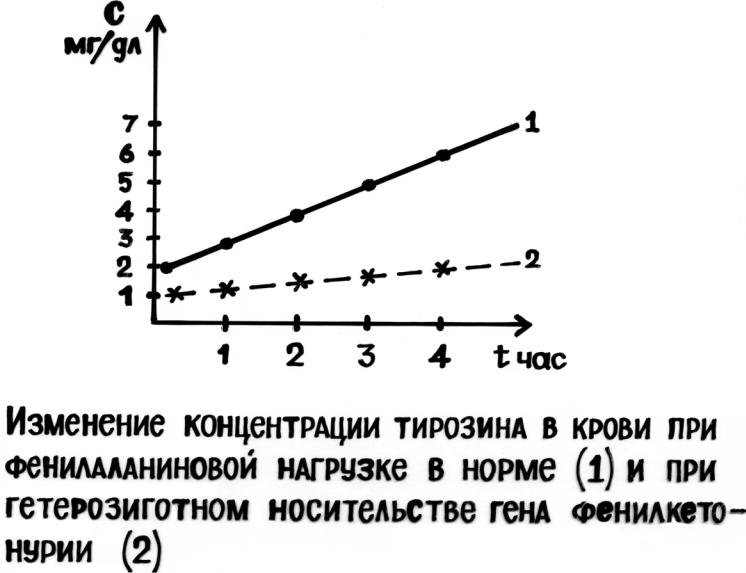
Для диагностики фенилкетонурии определяют концентрацию фенилаланина и патологических метаболитов в крови и моче больного.
В генетической консультации, для выявления гетерозиготного носителя заболевания, используют тест толерантности к фенилаланину:
Разработаны специальные схемы скрининга для выявления новорождённых детей с ФКУ.
В настоящее время для выявления мутантного гена фенилаланингидроксилазы у гетерозиготных носителей ФКУ, используют также ПЦРдиагностику.

Особенности обмена тирозина в разных тканях
Кроме использования в синтезе белков, Тир в разных тканях используется для синтеза многих биологически-активных соединений.
Катаболизм Тир до конечных продуктов происходит в печени.

Катаболизм Тирозина в печени:
В печени происходит катаболизм Тир до конечных продуктов:

Ферменты: 1 – тирозинаминотрансфераза (кофермент: ПФ);
2 – п-гидроксифенилпируватдиоксигеназа (кофакторы: вит. C и Fe2+);
3 – диоксигеназа гомогентизиновой кислоты (кофакторы: вит. C и Fe2+);
4 – фумарилацетоацетатгидролаза.
При наследственном дефекте гена фермента
диоксигеназы гомогентизиновой кислоты возникает заболевание – Алкаптонурия («чёрная моча»).
При этом заболевании с мочой выделяется большое количество гомогентизиновой кислоты.
При её окислении O2 воздуха образуются алкаптоны – темные пигменты.
Симптомы: 1) Моча приобретает черный цвет (у грудных детей – темные пятна на пеленках);
2)Охронозы – черные пятна в хрящах. (Очень часто: на кончике носа и мочках ушей);
3)Артриты – из-за отложения алкаптонов в суставах.
Частота заболевания: 2-5 случаев на ~ 1 млн. новорожденных.
Заболевание наследуется по аутосомнорецессивному типу.
Диагностических методов выявления гетерозиготных носителей – не найдено.
Превращение тирозина в меланоцитах.
В меланоцитах (пигментных клетках) Тир превращается в темные пигменты – меланины:
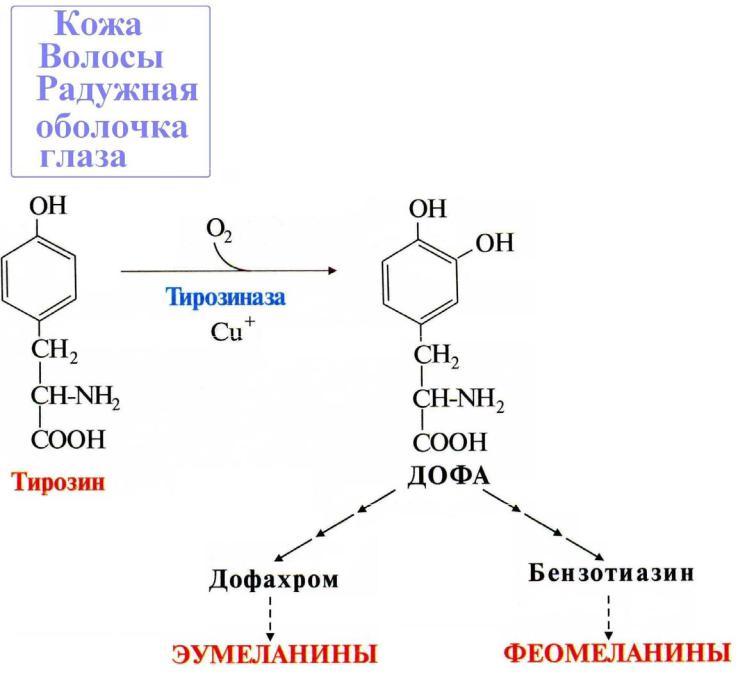
При наследственном дефекте гена фермента тирозиназы возникает заболевание – Альбинизм.
Симптомы: отсутствие пигментации кожи и волос. У больных часто снижена острота зрения, возникает светобоязнь.
Длительное пребывание под открытым солнцем приводит к раку кожи.
Частота заболевания: 1 случай на ~ 20 000 человек.
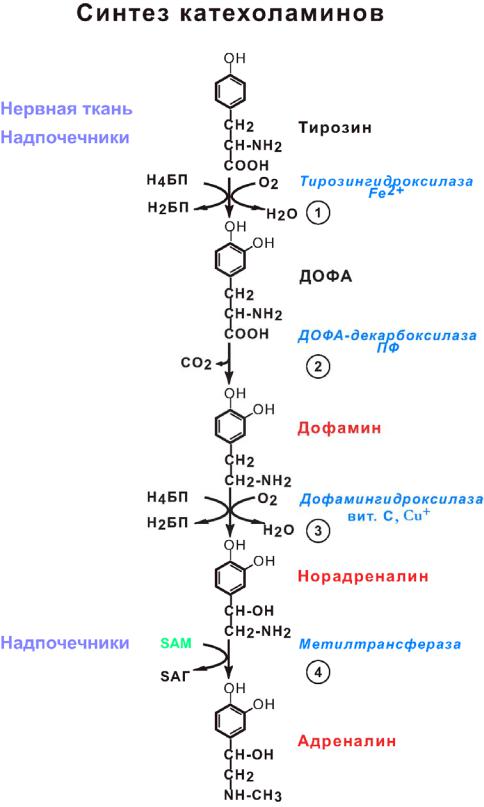
Превращение тирозина в надпочечниках и нервной ткани (синтез катехоламинов).
В мозговом веществе надпочечников и нервной ткани из тирозина синтезируются катехоламины (дофамин, норадреналин и адреналин):
Дофамин и норадреналин – нейромедиаторы разных отделов головного мозга.
Участвуют в синаптической передаче нервных импульсов.
Адреналин – гормон широкого спектра действия, регулирующий энергетический обмен, работу сердечно-сосудистой системы и др.
С нарушением синтеза катехоламинов связано несколько заболеваний:
1. Болезнь Паркинсона (Паркинсонизм)
Возникает из-за недостаточности дофамина в черной субстанции мозга.
Причины: снижение активности фермента ДОФАдекарбоксилазы, реже – тирозингидроксилазы.
Основные симптомы: тремор (непроизвольное дрожание), акинезия (скованность движений), ригидность (напряжение мышц).