

практические работы по кинетике
.pdfРис. 2. (а) Изменение концентрации реагента А (1), промежуточного вещества В (2) и конечного продукта С (3) по мере протекания параллельной реакции.
(б)Изменение значений максимальной концентрации промежуточного вещества Вмах
сростом соотношения констант скоростей стадий k2/k1
Рассмотрим пример использования метода Боденштейна для описания кинетики реакции разложения силана. Суммарную реакцию разложения силана с образованием кремния и водорода можно записать следующим образом:
SiH4 Si + 2 H2.
Механизм реакции состоит из 3-х стадий:
1) SiH4 |
SiH2 + H2 |
(k1) – константа скорости стадии 1 |
2) SiH2 |
Si + H2 (k2) – константа скорости стадии 2 |
|
3) SiH2 |
+ H2 SiH4 |
(k3) – константа скорости стадии 3 |
Выведем уравнение для скорости суммарной реакции разложения силана:
d[SiH4 ] k1[SiH4 ] k3[SiH2 ][H2 ] . dt
В этом уравнении присутствует концентрация промежуточного вещества
– SiH2, надежно измерить которую в ходе реакции чрезвычайно сложно. Поэтому, используя принцип стационарных концентраций, полагая концентрацию промежуточного соединения (в силу его высокой реакционной способности) малой и постоянной, запишем скорость ее изменения во время процесса разложения силана:
d[SiH2 ] k1[SiH4 ] k2[SiH 2 ] k3[SiH 2 ][H 2 ] 0. dt
Выразим из полученного соотношения концентрацию промежуточного соединения:
11
[SiH2 ] |
k1[SiH4 ] |
|
. |
|
k2 k3[H2 ] |
||||
|
|
|||
Подставим полученное выражение в суммарное уравнение скорости разложения силана:
d[SiH4 ] |
k1[SiH4 |
] k3k1[SiH4 ][H2 ] . |
dt |
|
k2 k3[H 2 ] |
После алгебраических преобразований выражение для скорости разложения силана запишется следующим образом:
|
d[SiH4 ] |
|
k1k2[SiH4 ] |
|
dt |
|
. |
||
k2 k3[H2 ] |
||||
Из полученного кинетического уравнения видно, что увеличение концентрации водорода в реакционной смеси будет приводить к снижению общей скорости разложения силана.
1.7. Энергия активации
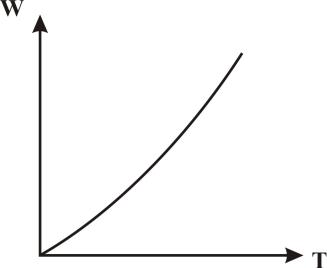
Для большинства химических реакций (за исключением тримолекулярных и ферментативных) скорость реакции увеличивается с ростом температуры. На рис. 3 дан примерный вид этой зависимости. Анализ кинетического
уравнения: W kcAn11 cAn22 показывает, что изменение скорости реакции с темпе-
ратурой может быть связано с изменением трех величин: константы скорости, концентрации реагентов и порядка реакций. Однако влияние температуры на концентрацию и порядок невелико; и в химической кинетике изменение скорости реакции от температуры связывают, в первую очередь, с изменением константы скорости реакции.
Рис. 3. Зависимость скорости химической реакции от температуры
Представление о зависимости скорости химической реакции от температуры дается эмпирическим правилом Вант-Гоффа: при повышении температу-
12
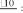
ры на десять градусов скорость реакции увеличивается в 2–4 раза. Правило действует в узком интервале температур, близких к комнатной.
Температурный коэффициент скорости реакции KT 10 позволяет при-
KT
ближенно оценить изменение скорости реакции при увеличении температуры на некоторое число градусов, равное (10·m), где значение коэффициента m может быть целым и дробным:
W2 |
|
KT m |
m . |
|
KT |
||||
W1 |
|
|
Количественная зависимость константы скорости реакции от температуры дается уравнением Аррениуса, которое в дифференциальной форме выглядит так:
d ln k |
|
Ea |
, |
(12) |
dT |
RT 2 |
где Еа – энергия активации.
Превращения исходных частиц в продукты реакции, как правило, связано с преодолением потенциального барьера. Его наличие обусловлено тем, что перестройка реагирующих частиц требует разрыва или ослабления отдельных химических связей, на что необходимо затратить энергию.
Другими словами, чтобы прореагировать, молекула должна иметь внутреннюю энергию, превышающую некоторую пороговую величину Еa. Доля частиц с энергией больше Еa равна exp(–Ea/RT) (закон Больцмана). В химическом превращении участвуют только такие частицы, энергия которых больше Ea, поэтому константа скорости пропорциональна exp(–Ea /RT).
Тогда интегрирование уравнения (12) в неопределенных пределах с последующим потенцированием дает следующее выражение (уравнение Аррениуса):
k A e |
Ea |
|
ln k ln A |
Ea |
|
|
RT |
или |
(12а). |
||||
RT |
||||||
|
|
|
|
|
В это выражение входят два параметра: энергия активации – Еа и предэкспоненциальный множитель (предэкспонента) – А.
Химическая реакция подчиняется теории Аррениуса, если выполнены следующие условия:
1)активация молекул происходит только за счет тепловой энергии (если молекулы возбуждаются светом, заряженными частицами и т.д., то закон Аррениуса может не выполняться);
2)протекание реакции не должно нарушать равновесного распределения энергии в системе.
Энергия активации – это избыток энергии по сравнению со средней
энергией реагирующих молекул при данной температуре, необходимый для того, чтобы произошла химическая реакция между этими частицами. Величина энергии активации зависит от вида реагирующих частиц (молекулы, радикалы, атомы, ионы и т.д.) и от их энергетического состояния.
13
Для реакций между валентно-насыщенными молекулами энергия активаций близка к энергии диссоциации и составляет 200–800 кДж/моль. Реакции между атомами и радикалами происходят с энергией активации, близкой к нулю, реакции атомов (радикалов) с молекулами протекают с промежуточными значениями энергии активации.
Для сложных реакций Еа, как правило, эффективная величина, представляющая собой алгебраическую сумму энергий активаций отдельных стадий. Это так называемая эффективная энергия активации – Еэфф.
Наблюдаются случаи, когда Eэфф < 0 (отрицательна). Например, для реакций типа:
А+ В С продукты
вслучае быстрого установления равновесия Еэфф < 0. Для случая, когда первая
стадия экзотермична, и по абсолютной величине Н (тепловой эффект первой стадии) > Еэфф.
Расчет энергии активации
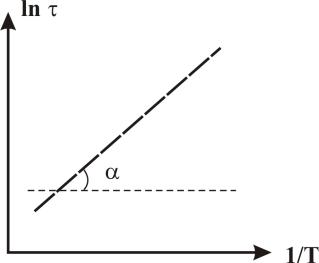
1. По температурной зависимости константы, скорости реакции с использованием выражений (12а) (рис. 4).
ln k ln A ERa T1
Ea tg( ) R
Рис. 4. Графическое представление уравнения Аррениуса (12а)
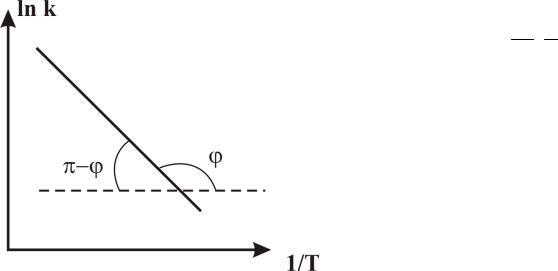
2. По температурной зависимости скорости реакции при постоянной концентрации реагентов (рис. 5). (Метод начальных скоростей реакций).
14
W k CA CB A e Ea RT CA CB CA const
CB const
lnW ln(A CA CB ) RTEa
Ea tg( ) R
Рис. 5. Определение энергии активации по зависимости скорости реакции от пературы
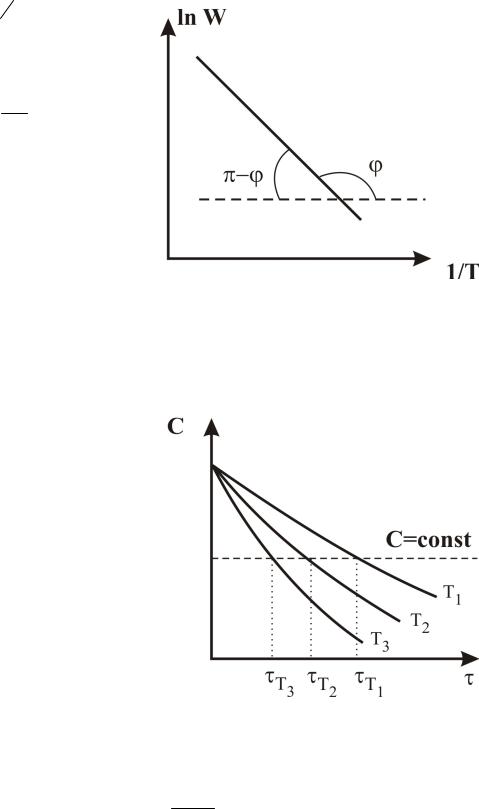
3. При изучении кинетики реакций в растворах часто вычисляют энергию активации не из температурной зависимости констант скоростей, а из температурной зависимости времени протекания реакции на одну и ту же глубину. На рис. 6 представлены кинетические кривые, полученные для различных температур Т1, Т2, Т3. [7].
Рис. 6. Определение энергии активации методом равно-процентных выходов или методом полупревращений
Вместо уравнения |
Ea RT 2 d ln k |
для расчета Еа можно исполь- |
зовать уравнение вида: |
dT |
|
|
|
Ea RT 2 (ddTln ) ,
где индекс ( ) показывает одну и ту же концентрацию реагента в смеси при разных температурах.
Для определения энергии активации с помощью уравнения (12) необходимо точно измерить время достижения одной и той же степени превращения при разных температурах. После интегрирования уравнения (12):
15
Ea tg К
ln const |
Ea |
(12б) |
RT |
|
Рис. 7. Определение энергии активации по уравнению (12б)
Для расчёта энергии активации по методу (3) необходимо убедиться в сохранении одной и той же кинетической зависимости во всем интервале температур.
16
2. КАТАЛИЗ И ОСНОВНЫЕ ПОНЯТИЯ
Явление катализа можно определить как изменение скорости химической реакции под влиянием веществ – катализаторов, многократно вступающих в промежуточное химическое взаимодействие с участниками реакции и восстанавливающих свой химический состав после каждого цикла промежуточных взаимодействий [9].
Катализатор не может вызвать реакцию, которая термодинамически запрещена, из определения следует, что он изменяет лишь кинетические параметры реакции. Особенно важно, что при катализе происходит промежуточное химическое взаимодействие катализатора с реагирующими веществами. Этим подчеркивается химическая сущность катализа и проводится четкая граница между каталитическими процессами и явлениями изменения скорости химических реакций под влиянием различных физических факторов, (Например, инертные насадки увеличивают скорость поглощения газа жидкостью за счет увеличения поверхности контакта, но эти процессы, конечно, не могут быть отнесены к каталитическим).
Катализатор выходит из реакции химически неизменным, и следствием этого является неспособность катализатора изменить положение химического равновесия.
Следствием многократности участия катализатора в элементарных стадиях процесса является способность катализаторов, взятых в очень небольших количествах, превращать, большие массы исходных веществ.
Приведенное определение четко ограничивает круг явлений, которые могут быть отнесены к каталитическим, однако, их количество и разнообразие требует дополнительной классификации внутри понятия катализа.
Наиболее простая система классификации каталитических процессов основана на фазовом принципе.
Гомогенный катализ. К этой группе относятся каталитические реакции, в которых реагирующие молекулы, и катализатор в форме атомов, молекул или ионов, находятся в одной фазе и образуют гомогенную химическую систему.
Гетерогенный катализ. В гетерогенных каталитических процессах реагирующие вещества и катализатор находятся в разных фазах, и реакция протекает на границе раздела фаз.
Ферментативный (или микрогетерогенный) катализ. Относящиеся к этой группе процессы занимают промежуточное положение между первыми двумя группами. Это каталитические коллоидные системы, в которых реагирующие вещества находятся в растворе, а катализатор в коллоидно-дисперсном состоянии, главным образом, это процессы, ускоряемые ферментами. В этих системах к чертам гомогенного катализа в растворе добавляются черты гетерогенного катализа вследствие большого числа атомов, входящих в частицы катализатора.
Существенным недостатком классификации по фазовому принципу является игнорирование особенностей взаимодействия катализатора с реагирующи-
17
ми веществами, т.е. механизма промежуточного взаимодействия. Хотя в настоящее время мы не имеем достаточного количества надежных данных о глубоком механизме каталитических реакций, однако разделение на два основных класса (слитный и раздельный механизмы катализа) может быть сделано, на основе классификации, принятой в химической кинетике.
Химические реакции сводятся к разрыву определенных старых связей в молекулах реагирующих веществ и образованию новых. Наиболее распространенным видом связи в химических соединениях является двухэлектронная связь. Разрыв ее может приводить к разрушению электронной пары с образованием неспаренных электронов у каждого из разделяемых атомов (гомолитические реакции) или к переходу электронной пары без разрушения к одному из атомов (гетеролитические реакции). Применяя этот подход к процессу взаимодействия реагирующих веществ с катализатором, можно выделить гомолитический катализ, когда про межуточное химическое взаимодействие протекает по гомолитическому механизму, и гетеролитический катализ, если это взаимодействие имеет гетеролитическую природу.
По гомолитическому механизму протекают реакции окислительновосстановительного типа: гидрирования, дегидрирования, окисления, восстановления, синтеза аммиака и др. Катализаторы этих процессов должны поставлять неспаренные электроны для образования новых электронных пар, поэтому активными здесь являются переходные элементы в виде металлов, оксидов, сульфидов и т.п.
Гетеролитический механизм наблюдается при каталитических реакциях дегидратации спиртов, гидратации олефинов, крекинг, изомеризации и др. Катализаторы этих процессов должны обладать способностью к образованию координационной связи путем отдачи или присоединения электронной пары. В частности, они могут быть протонными или апротонными кислотами и основаниями.
2.1.Гомогенные каталитические реакции
2.1.1.Кинетика гомогенных каталитических реакций
Большинство каталитических процессов протекает через ряд последовательных стадий. Действие положительных катализаторов заключается в открытии нового реакционного пути, благодаря промежуточному химическому взаимодействию реагирующих веществ с катализатором. Этот путь характеризуется более низкими значениями энергии Гиббса образования активированных комплексов всех стадий. На этом пути (рис. 8) возможно образование одного (пунктир) или нескольких стабильных промежуточных соединений, отвечающих минимумам энергии Гиббса. Однако стадийность сама по себе не приводит к увеличению скорости реакции, если при этом не снижаются абсолютные высоты всех энергетических, барьеров, определяемых от уровня энергии исходных веществ.
18
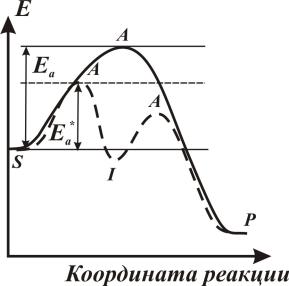
Часто общая скорость процесса определяется лимитирующей стадией, а на стадиях, которые не лимитирующие, устанавливается квазиравновесное состояние.
Рис. 8. Активационный барьер для одностадийной (сплошная кривая) и двухстадийной (пунктир) химической реакции:
S – исходные реагенты;
Р– продукты реакции;
А– активированный комплекс;
I – интермедиат (промежуточное соединение)
Из рис. 8 видно, что энергия активации двухстадийной реакции Ea* существенно ниже энергетического барьера одностадийной реакции Еа, что обеспечивает увеличение скорости двухстадийной реакции по сравнению с одностадийной.
Можно выделить два принципиальных механизма каталитических реакций: слитный и стадийный. Слитный механизм (I) может быть представлен схемой бимолекулярной
А + К {А—К}* В + К
или тримолекулярной реакции: |
|
|
|
|
|
А1 + А2 |
+ К {А1—А2—К}* В1 |
+ В2 |
+ К. |
||
Стадийный (двухстадийный, например) механизм (П) может быть пред- |
|||||
ставлен: |
|
|
|
|
|
|
A |
k1 |
|
|
|
|
K A K |
, |
|
|
|
|
1 |
1 |
|
|
|
|
|
k 1 |
|
|
|
A1K A2 k2 продукты K
(в обоих случаях запись переходного состояния для краткости опущена).
Часто гомогенные каталитические реакции в растворах протекают по схеме:
Стадия 1. Образование промежуточного продукта АК в результате обратимого взаимодействие между катализатором и одним из исходных веществ.
Стадия 2. Взаимодействие этого промежуточного продукта со вторым исходным веществом.
Пусть первая стадия протекает с высокими скоростями и в ней устанавливается квазиравновесное состояние. Вторая стадия течет медленно и лимитирует скорость всего процесса. Тогда
19
W k2[ A1K ][A2 ]. |
(13) |
Из константы равновесия первой стадии концентрация промежуточного вещества выражается следующим образом:
[A1K] = K [A1] [K].
Вещество А1К, образующееся в результате стадии 1, называется промежуточным веществом Аррениуса.
Пусть теперь скорость второй стации велика, а k1 > k-1. Тогда на первой стадии не устанавливается квазиравновесное состояние, и она определяет, скорость всего процесса в целом:
W k1 [A] [K]. |
(14) |
Обычно в таких реакциях концентрация промежуточного вещества [A1K] очень мала, много меньше концентрации А1 и А2. Это – промежуточное вещество Вант-Гоффа. Как видно из уравнения (14), скорость реакции не зависит от концентрации А2, но это отклонение от закона действующих масс – кажущееся, поэтому на каждой из стадий он соблюдается, а уравнение (14) – суммарное.
2.1.2. Некоторые особенности протолитического взаимодействия
Из большого числа процессов гомогенного катализа в растворах наиболее полно изучены гетеролитические реакции. Важнейшим классом этих реакции является кислотно-основной катализ, включающий много численные процесса изомеризации, гидратации, дегидратации, гидролиза, алкилирования, этерификации и другие. В основе механизма этого класса каталитических реакций лежит промежуточное протолитическое взаимодействие реагирующих веществ с катализатором:
S + AH SH+ + A–.
Здесь S – реагирующее вещество,
АН – катализатор кислотной природы.
В процессе протолитического взаимодействия образуется комплекс, обладающий повышенной реакционной способностью в отношении многих реакций. Это связано с сильным поляризующим влиянием иона водорода, приводящим к значительным смещениям электронов внутри молекулы.
Протолитическое взаимодействие происходит и при катализе основаниями, например, по схеме:
SH + B BH+ + S–.
Здесь SН – реагирующее вещество, В – катализатор основной природы.
Одним из факторов, определяющих сравнительную активность кислот и оснований (как катализаторов гомогенных реакций), является их сила. Можно предположить, что чем сильнее кислота, тем легче (быстрее) она передает протон реагирующему веществу. Аналогично при катализе основаниями можно
20