

Истинная полицитемия. Капустинский М.Н
..pdf«ИСТИННАЯ ПОЛИЦИТЕМИЯ»
ИСТИННАЯ ПОЛИЦИТЕМИЯ.
Истинная полицитемия (болезнь Вакеза, эритремия) – хроническое неопластическое миелопролиферативное заболевание с поражением стволовой клетки, пролиферацией 3 ростков кроветворения, повышенным образованием эритроцитов и в меньшей степени лейкоцитов и тромбоцитов.
На определенном этапе, а иногда и с самого начала к пролиферации гемопоэтических клеток в костном мозге присоединяется миелоидная метаплазия селезенки.
Распространенность: 0,6-1,6 на 100000 населения. Не являются редкостью случаи заболеваний в молодом возрасте. У молодых людей заболевание протекает более неблагоприятно.
Соотношение мужчины/женщины равно 1,2:1.
Этиология неизвестна.
Значение генетической предрасположенности подтверждают случаи семейной полицитемии. Частота семейной полицитемии составляет 0,38 на 100 больных истинной полицитемией (ИП).
Патогенез.
Клональное, неопластическое происхождение ИП было доказано наличием одного типа Г-6-ФДГ во всех клетках миелоидного ряда у гетерозиготных по этому ферменту женщин-мулаток, больных ИП: в эритроцитах, гранулоцитах и тромбоцитах обнаружен только один тип, тогда как в фибробластах кожи и костного мозга – оба типа этого фермента.
Цитогенетика.
Специфических цитогенетических аномалий при ИП не найдено.
В 25,4% - хромосомные аберрации в 20, 8, 9, 19 паре, изредка в 13 и 12 хромосоме. Самая частая патология – это +9р (включая +9 трисомию и тетрасомию 9р). (А. В. Демидова, 2007, Y. T. Prehal, 2001).
Морфологических, ферментных и цитогенетических признаков поражения лимфатической системы при эритремии не имеется, однако функциональное состояние Т-лимфоцитов изменено: обнаружен сниженный ответ на известные мутагены и повышение их спонтанной активности, снижена способность Тлимфоцитов к синтезу ИЛ - 2.
Патологическая анатомия.
Состояние костного мозга при ИП характеризуется гиперплазией трех или двух ростков кроветворения. Следует различать:
1.Классический вариант, который обозначается панмиелозом: тотальная гиперплазия трех ростков кроветворения с выраженным мегакариоцитозом и редукцией жировой костной ткани вплоть до ее полного отсутствия; мегакариоциты отличаются крупными размерами.
2.Гиперплазию эритроидного и гранулоцитарного ростков с
небольшой степенью мегакариоцитоза.
3.Гиперплазию эритроидного и мегакариоцитарного ростков.
4.Пролиферацию одного эритроидного ростка.
Клиническая картина.
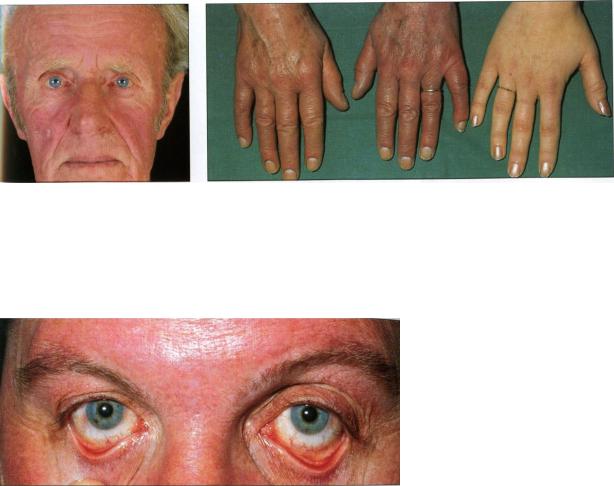
Заболевание начинается постепенно. Нарастают покраснение кожных покровов, характерна эритроцианотическая окраска кожи кистей рук и лица (рис 1а,б, 2), видимых слизистых оболочек, особенно мягкого неба, резко контрастирующая с обычной окраской твердого неба (симптом Купермана). Конечности горячи на ощупь. Больные испытывают чувство жара, плохо переносят жару. Кожные покровы обычно чистые, но нередко возникает трудно квалифицируемый дерматит лица пустулезного типа. Также появляется слабость, тяжесть в голове, увеличение селезенки, артериальная гипертония, а у половины больных - мучительный кожный зуд после умывания, мытья, плавания. Иногда первыми проявлениями заболевания становятся некрозы пальцев нижних конечностей, тромбозы более крупных артерий нижних и верхних конечностей, носовые кровотечения, тромбофлебит, инфаркт миокарда или легкого и, особенно, эритромелалгия - острые жгучие боли в кончиках пальцев, устраняемые ацетилсалициловой кислотой на 1-3 дня.
Рис 1а. Плетора у 65-летнего |
1б. Эритремия. Кисти 50-летней больной |
больного. Нв-220 г/л, L-17×109/л, |
(Нв-200 г/л, L-15×109/л, Tr-550×109/л). |
Tr-550×109/л, ОЦЭ – 65 мл/кг |
Показанные слева выглядят отечными и (по В. |
Хоффбранд, |
полно-кровными по сравнению с кистью |
Дж. Петит, 2007) |
здоровой женщины (Нв-145г/л) |
|
(по В. Хоффбранд, Дж. Петит, 2007) |
Рис 2. Полнокровие лица и гиперемия коньюнктив у 40-летней больной. (по В. Хоффбранд, Дж. Петит, 2007)
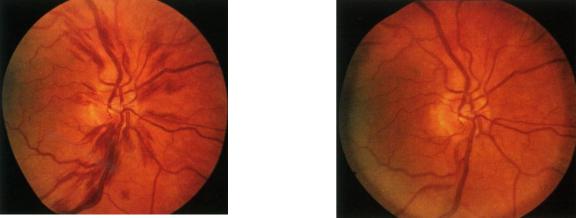
Увеличение массы циркулирующих эритроцитов и показателя гематокрита приводит к повышению вязкости крови, замедлению тока крови, стазам крови на уровне микроциркуляции, повышению периферического сосудистого сопротивления. Этим объясняется высокая частота сосудистых, преимущественно церебральных жалоб. Иногда они носят характер мучительной мигрени с нарушением зрения (рис 3а, б).
Рис 3а. Проявления синдрома повышенной |
Рис 3б. Тот же случай, после |
вязкости крови: сильное расширение |
кровопускания - исчезли |
сосудов сетчатки, кровоизлияния в |
кровоизлияния и отек сетчатку и легкий |
отек диска зрительного нерва. |
зрительного нерва, |
Больной жаловался на головную боль, |
нормализовался диаметр |
вялость, спутанность сознания и |
сосудов. |
нечеткость зрения. |
(по В. Хоффбранд, Дж. Петит, 2007) |
(по В. Хоффбранд, Дж. Петит, 2007) |
|
Вто же время у многих больных имеется удивительная приспособляемость к плеторе и отсутствуют жалобы. При этом риск острых нарушений мозгового кровообращения сохраняется.
Ванамнезе у многих больных задолго до установления диагноза имеются указания на кровотечение после экстракции зубов, "хорошие" (т.е. несколько повышенные) показатели красной крови, которым не придавали должного значения.
Артериальная гипертензия наблюдается у 35-40 % больных на момент постановки диагноза. Различают: симптоматическую (плеторическую) артериальную гипертензию, причинно связанную с повышением вязкости крови; сопутствующую эссенциальную, отягощенную плеторой; вазоренальную, обусловленную склеротическим или тромботическим стенозом почечных артерий.
Клинические симптомы ИП условно можно разделить на:
1)обусловленные увеличением массы циркулирующих эритроцитов, т.е. плеторой;
2) вызванные пролиферацией тромбоцитов и гранулоцитов. Они зависят от стадии заболевания.
Стадии заболевания.
ВI стадии, продолжительность которой составляет 5 лет и более, наблюдается умеренная плетора, селезенка не пальпируется. В крови на этой стадии преобладает умеренный эритроцитоз. В костном мозге - картина панмиелоза. Сосудистые и висцеральные осложнения в это время возможны, но не часты.
II А стадия процесса - эритремическая - является развернутой, для нее не характерна миелоидная метаплазия селезенки. Продолжительность этой стадии составляет 10-15 лет и более. Субъективное состояние в это время чаще нарушено. Выражены плеторический синдром, спленомегалия, а несколько ранее - и гепатомегалия. Тромбозы артериальных и венозных сосудов, геморрагические осложнения на этой стадии наблюдаются чаще, чем в I ст. Анализ крови указывает на "чистую" эритроцитемию или эритроцитемию и тромбоцитоз или панмиелоз и нейтрофилез с палочкоядерным сдвигом, увеличением числа базофилов. В костном мозге наблюдается тотальная трехростковая гиперплазия с выраженным мегакариоцитозом, возможны ретикулиновый и очаговый коллагеновый миелофиброз.
Причиной спленомегалии в стадии IIА является усиление депонирования и секвестрации форменных элементов крови, доказанное многочисленными исследованиями. Степень увеличения селезенки в стадии IIА небольшая или умеренная. Значительные размеры селезенки обусловлены осложнением – портальной гипертензией. Это подтверждается изучением последствий случайных спленэктомий при нераспознанной ИП, после которой наблюдается резкое нарастание плеторического синдрома, лейкоцитоза и тромбоцитоза.
Ко II Б стадии также относится эритремический, развернутый процесс, но с миелоидной метаплазией селезенки. Плетора может быть выражена в большей или меньшей степени, наблюдается спленомегалия и гепатомегалия. В крови в этой стадии отмечается панцитоз с лейкоцитозом выше 15х103 (15000) в 1 мкл и сдвигом лейкоцитарной формулы до миелоцитов, единичные эритрокариоциты. В костном мозге, как и во IIА стадии, панмиелоз, но может преобладать гиперплазия гранулоцитарного ростка, возможен ретикулиновый и очаговый коллагеновый миелофиброз. В клинической картине нередко ведущими оказываются аллергические осложнения и уратовый диатез.
Вэтой стадии могут наблюдаться истощение больного, рецидивирующие тромботические осложнения и кровоточивость. В стадии IIБ спленомегалию вызывает прогрессирующее развитие миелоидной метаплазии. Ей соответствует появление левого сдвига в лейкоцитарной формуле, эритрокариоцитоза и морфологических изменений в эритроцитах: каплевидного пойкилоцитоза (tear-drop), анизоцитоза. В поздний период постэритремической миелоидной метаплазии появляются макроцитоз и другие морфологические признаки макроцитарных анемий, а также осколки
ядер мегакариоцитов. Картина периферической крови в этой стадии тождественна, наблюдаемой при идиопатическом миелофиброзе.
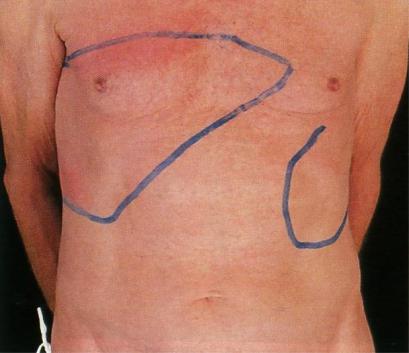
Увеличение печени часто сопутствует спленомегалии (рис. 4).
Рис 4. Истинная полицитемия. Увеличение печени и селезенки (по В. Хоффбранд, Дж. Петит, 2007).
Встадии IIА оно обусловлено повышенным кровенаполнением печени,
встадии IIБ – миелоидной метаплазией. Для обеих стадий характерно развитие фиброза печени, а также холелитиаза, причина которого заключается в чрезмерной густоте желчи. Наблюдается цирроз печени как осложнение ИП или обусловленный сопутствующим хроническим гепатитом.
III стадию эритремии называют анемической или постэритремической
миелоидной метаплазии с миелофиброзом или без. Бывают и анемический и тромбоцитопенический синдромы или даже панцитопения. В костном мозге может быть выражен миелофиброз, миелопоэз в одних случаях сохранен, а в других редуцирован. В увеличенных селезенке и печени наблюдается миелоидная метаплазия.
С развитием эритремии (III стадия), нередко наблюдается дефицит железа, нивелирующий плетору. Клинические проявления дефицита железа чаще наблюдаются у лиц старшего возраста.
Развитию анемической стадии предшествует определенная динамика клинико-лабораторных данных, в частности, увеличение селезенки, постепенное уменьшение плеторического синдрома, появление лейкоэритробластической картины периферической крови. В костном мозге постепенно развивается миелофиброз, которому могут сопутствовать смена типа клеточной пролиферации, нарастание патологии сосудов костного мозга и неэффективность эритропоэза - исход эритремии во вторичный миелофиброз.
Нарушение обмена мочевой кислоты (гиперурикемия и урикозурия),
свойственное всем хроническим миелопролиферативным заболеваниям (ХМПЗ), становится особенно частым и клинически манифестным в стадиях
IIБ и III. Клинические проявления уратового диатеза — это почечная колика,
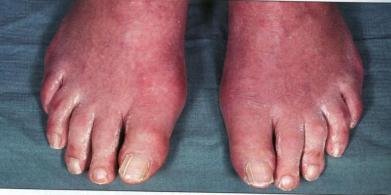
подагра, подагрическая полиартралгия или их сочетание (рис 5).
Рис 5. Истинная полицитемия. Подагра: воспаление и отек плюснефалангового и межфалангового суставов большого пальца правой стопы. (по В. Хоффбранд, Дж. Петит,
2007).
Отмечается клиническое своеобразие течения подагры и подагрической полиартралгии. Нефролитиаз иногда становится главной клинической проблемой, приводит к обструкции мочеточников, вызывает осложнение хроническим пиелонефритом. В дальнейшем развивается острая почечная недостаточность и хроническая болезнь почек. Циторедуктивная терапия в период ее проведения может повысить степень гиперурикемии, поэтому всегда должна сопровождаться приемом аллопуринола.
Причиной нарушения уратового обмена при всех ХМПЗ, в том числе ИП, является повышенный эндогенный синтез мочевой кислоты, вызванный клеточным гиперкатаболизмом и частично неэффективным эритропоэзом.
Частыми осложнениями развернутой стадии заболевания являются микроциркуляторные расстройства с клиникой эритромелалгии, преходящих нарушений церебрального и коронарного кровообращения и геморрагических отеков голеней, а также тромбозы венозных и артериальных сосудов и кровотечения.
К висцеральным осложнениям ИП относят язвы желудка и двенадцатиперстной кишки, частота которых составляет 10—17 %. Помимо язв, при ИП почти постоянно выявляют гиперемию слизистой оболочки, а также эрозии желудка и двенадцатиперстной кишки. Это осложнение вызывается плеторическим кровообращением, гистаминемией, серотонинемией. Часто больные по тем или иным причинам принимают ацетилсалициловую кислоту, которая также оказывает ульцерогенное воздействие и способствует возникновению кровотечения. Адекватная терапия основного заболевания приводит к исчезновению клинических проявлений язвенного поражения без дополнительного назначения противоязвенной терапии, которая сама по себе у больных ИП малоэффективна.
Патогенез тромботических осложнений истинной полицитемии состоит в увеличении массы циркулирующих эритроцитов, замедлении тока крови и повышении ее вязкости. Их развитию способствуют тромбоцитоз и качественные нарушения тромбоцитов. В плазме крови нередко определяются циркулирующие агрегаты тромбоцитов, что бывает следствием не только их количественного увеличения, но и нарушения функциональных свойств тромбоцитов.
Сосудистые осложнения являются главной опасностью для здоровья и жизни больных. Различают:
1.Микрососудистые тромбофилические осложнения с клиническими проявлениями в виде эритромелалгии, головной боли, преходящих нарушений зрения, стенокардии и др.
2.Тромбозы артериальных и венозных сосудов, локальные и множественные. Тромбофилические осложнения ИП обусловливаются увеличением массы циркулирующих эритроцитов (МЦЭ), тромбоцитозом, лейкоцитозом, т.е. первичными нарушениями клеточного гемостаза. Увеличение МЦЭ приводит к нарушению реологии крови, повышению ее вязкости, замедлению капиллярного кровотока, стазам крови на уровне микрососудов, сближению клеток между собой, усилению контакта клеток с сосудистым эндотелием и нарушению суспензионной способности эритроцитов.
3.Геморрагии и кровотечения, спонтанные и спровоцированные любыми, даже малыми оперативными вмешательствами.
4.ДВС-синдром с клиническими проявлениями в виде локальных и множественных тромбозов и кровотечений (тромбогеморрагический синдром).
Одновременная склонность как к тромбозам, так и к кровотечениям -
уникальная особенность истинной полицитемии.
Кровотечения, спонтанные и спровоцированные оперативными вмешательствами, обусловлены нарушениями свертывания крови, фибринолиза и ретракции кровяного сгустка. Патогенез микроциркуляторной кровоточивости определяется преимущественно снижением агрегации тромбоцитов.
Геморрагический синдром при ИП проявляется спонтанной кровоточивостью десен, носовыми кровотечениями и экхимозами, характерными для нарушений первичного, тромбоцитарно-сосудистого гемостаза. В то же время при оперативных вмешательствах, даже таких малых, как экстракция зуба, возникают массивные кровотечения, что возможно при нарушениях в системе свертывания крови и фибринолиза.
Геморрагические осложнения истинной полицитемии полностью ликвидируются у больных, леченных кровопусканиями, когда нормализуется показатель гематокрита.
Критерии диагностики ИП (ВОЗ), изложенные в Классификации опухолей (World Health Organisation Classification of Tumors, 2001).
К категории А отнесены следующие:
А1 — увеличение массы циркулирующих эритроцитов (МЦЭ) на 25 % по сравнению со средней нормой или увеличение уровня гемоглобина (НЬ) более 185 г/л у мужчин и 165 г/л у женщин;
А2 — отсутствие причин для вторичного эритроцитоза (включая фамильный эритроцитоз, гипоксию, высокое сродство гемоглобина к кислороду, врожденные мутации эритропоэтинового рецептора, повышенную продукцию эритропоэтина опухолями);
A3 — наличие спленомегалии;
А4 — наличие клональных хромосомных аберраций (исключая обнаружение Ph-хромосомы или слитного гена BCR/ABL);
А5 — рост эритроидных колоний in vitro без добавления эритропоэтина.
К категории В относятся:
В1 — увеличение количества тромбоцитов более 400 • 109/л; В2 — количество лейкоцитов более 12 • 109/л;
В3 — обнаружение панмиелоза с увеличением пролиферации эритробластов и мегакариоцитов в трепанате костного мозга;
В4 — низкий уровень эндогенного эритропоэтина в сыворотке крови. Диагноз ИП считается достоверным, если имеется сочетание признаков
А1+А2+ любой другой из категории А или А1+А2 + любые 2 из категории В.
Т.о Основными диагностическими критериями ИП являются:
♦высокие значения МЦЭ;
♦наличие спленомегалии;
♦низкое содержание эритропоэтина, отсутствие или малая степень его изменений в ответ на кровопускание;
♦характерен спонтанный рост эритроидной культуры без добавления эритропоэтина;
♦картина панмиелоза (характерна трехростковая гиперплазия костного мозга, и важно обнаружение гиперплазии мегакариоцитов)
|
Дифференциальный |
диагноз: |
эритроцитозы, |
эссенциальная |
|
|
|
|
|
||
тромбоцитопения, хронический идиопатический миелофиброз. |
|
||||
|
Классификация абсолютных эритроцитозов |
||||
|
(по C. Pearson, M. Messinezy, N. Westwood, 2000). |
||||
Первичный эритроцитоз. |
|
|
|
|
|
Врожденный. |
|
|
|
|
|
– |
Мутация гена рецептора к эритропоэтину. |
|
|||
Приобретенный. |
|
|
|
|
|
– |
Истинная полицитемия. |
|
|
||
Вторичный эритроцитоз. |
|
|
|
|
|
Врожденный.
– |
Гемоглобинопатии с повышенным сродством к кислороду. |
– |
Автономная гиперпродукция эритропоэтина. |
Приобретенный. |
|
– |
Гипоксемия. |
– |
Болезни почек. |
Идиопатический эритроцитоз.
Причины вторичных эритроцитозов.
Врожденные.
|
Мутантный гемоглобин с повышенным сродством к кислороду. |
|
Врожденный дефицит 2,3 DPG (дифосфоглицерат) в |
эритроцитах. |
|
|
Автономное повышение продукции эритропоэтина. |
Приобретенные.
Артериальная гипоксемия.
Высотная болезнь.
Врожденные «синие» пороки сердца.
Хронические бронхолегочные заболевания.
Другие причины ухудшения обеспечения тканей кислородом.
Курение табака.
Болезни почек.
Опухоли почек.
Кисты почек.
Диффузные паренхиматозные заболевания.
Гидронефроз.
Стеноз почечных артерий.
Трансплантация почек.
Эндокринные повреждения.
Адреналовые опухоли.
Гемангиобластома мозжечка.
Фиброма матки.
Карцинома бронхов.
Лекарства.
Андрогены.
Заболевания печени.
Гепатома.
Цирроз.
Гипоксические эритроцитозы относятся к компенсаторным, а
паранеобластические и нефрогенные — к дисрегуляторным, поскольку они не выполняют физиологических функций. Все вторичные абсолютные эритроцитозы являются эритропоэтинзависимыми. Гипоксические и нефрогенные эритроцитозы возникают при усилении продукции эритропоэтинов (ЭП) почками в ответ на общую или локальную ишемию