

Множественная миелома. Капустинский М.Н
..pdf«МНОЖЕСТВЕННАЯ МИЕЛОМА»
МНОЖЕСТВЕННАЯ МИЕЛОМА.
Множественная миелома (ММ) - (миеломная болезнь, болезнь Рустицкого-Калера) – это злокачественное лимфопролиферативное заболевание, характеризующееся инфильтрацией костного мозга плазматическими клетками, наличием моноклонального иммуноглобулина в сыворотке крови и/или в моче и остеолитическими поражениями костей.
Обязательный симптом ММ - моноклональная иммуноглобулинопатия.
В соответствии с классификацией ВОЗ – ММ относится к периферическим В-клеточным лимфоидным опухолям.
ИСТОРИЯ ВОПРОСА.
►1844 – 1850 Первое описание клинического случая миеломы, названной “мягкие и хрупкие кости” (“mollities and fragilitas ossium”). Первому пациенту, Thomas Alexander McBean, диагноз был установлен в 1845 году доктором William Macintyre, консультантом Харлей-стрит в Лондоне. Необычные почечные проблемы, которые он выявил, были детально исследованы доктором Henry Bence Jones, который опубликовал свои
данные в 1848 году. В 1846 г. хирург John Dalrymple обратил внимание и опубликовал свои данные о том, что пораженные болезнью кости содержали клетки, которые, как впоследствии было показано, являются плазматическими клетками.
►Доктор Macintyre в 1850г. опубликовал статью, детально описав этот случай миеломы Бенса Джонса. Было обращено внимание, что доктором Samuel Solly был опубликован клинический случай миеломы (Sarah Newbury) еще в 1844г.
►1873 Rustizky ввел термин «множественная миелома», для того, чтобы указать на присутствие в костях множественных повреждений, вызванных плазматическими клетками.
►1889 Otto Kahler опубликовал подробное клиническое описание множественной миеломы, «болезни Кахлера».
►1890 Ramon y Cajal представил первое тщательное микроскопическое описание плазматической клетки.
►1900 Wright установил, что клетки множественной миеломы являются плазматическими клетками.
►1903 Weber обратил авимание на то, что миеломная костная болезнь (литические повреждения) выявляется при рентгенографии.
►1909 Weber предложил идею о том, что миеломную костную деструкцию вызывают костно-мозговые плазматические клетки.
►1930-е Рутинный диагноз миеломы оставался сложным до 30-х годов, когда впервые более широко стали использоваться костно-мозговые аспираты.
Развитие методик ультрацентрифугирования и электрофореза протеинов сыворотки/мочи улучшили как скринирование, так и диагностику.
►1953 Был введен иммуноэлектрофорез, который обеспечил точную идентификацию моноклональных миеломных протеинов. (Впоследствии была введена иммунофиксация как более чувствительный метод).
►1956 Korngold и Lipari обратили внимание на то, что протеины Бенса Джонса (BJ) имеют отношение к нормальному сывороточному гаммаглобулину, так же, как и аномальные сывороточные протеины. В их честь протеины Бенса Джонса были названы каппа (κ) и лямбда (λ).
►1958 Открытие сарколизина, из которого был получен мелфалан (Алкеран). Впервые стало возможным лечение миеломы.
►1961 Waldenstrom подчеркнул важность дифференцирования между моноклональными и поликлональными гаммопатиями. Он свызывал IgM-моноклональные протеины с макроглобулинемией, которая является отличным от миеломы состоянием.
►1962 Первое сообщение об успешном лечении миеломы мелфаланом (Алкераном),
сделанное Bergsagel.
►1964 Первое сообщение об успешном лечении миеломы циклофосфамидом
(цитоксаном), сделанное Korst. Результаты, полученные при лечении циклофосфамидом, соответствовали таковым при использовании мелфалана.
►1969 Сочетание мелфалана с преднизоном, как было показано Alexanian, обеспечило лучшие результаты по сравнению с монотерапией мелфаланом.
►1975 Введение системы стадирования миеломы Durie/Salmon. Пациенты классифицируются для того, чтобы определить эффект химиотерапии на разных стадиях заболевания ( I, II, IIIA или IIIB).
►1976 – 1992 Внедряются различные комбинации химиотерапевтических агентов, включая М2 режим (VBMCP), VMCP-VBAP и ABCM. Результаты лечения указывали на некоторое преимущество по сравнению с MP. Тем не менее, проведенный в 1992 сравнительный мета-анализ (Gregory) указал на эквивалентные результаты для всех комбинаций.
►1979 – 1980 Впервые был предложен индекс метки ( анализ растущей фракции) как тест при миеломе и родственных заболеваниях. Стабильная ремиссия или фаза плото при миеломе определяется как период, когда фракция роста миеломных клеток (L1%)
равна 0%.
►1982 Fefer и Osserman провели трансплантацию от близнеца в качестве лечения миеломы.
►1983 Bataille и Durie впервые использовали уровень сывороточного В2 микроглобулина в качестве прогностического теста.
►1984 Barlogi и Alexanian предложили схему химиотерапии VAD.
►1984 – 1986 Первые сообщения разных исследователей об аллогенных трансплантациях при ММ.
►1986 – 1996 Большое число исследований из разных центров, оценивающих высокодозную химиотерапю с последующей аутологичной трансплантацией костного мозга или клеток-предшественников периферической крови. Предложены процедуры одиночной (McElwain) и двойной (Barlogi) трансплантации.
►1996 Первое и до сих пор единственное рандомизированное исследование, указывающее на возможную пользу высокодозной химиотерапии с последующей ТКМ по сравнению со стандартной химиотерапией (Attal). До сих пор не было проведено метаанализа или какого-либо более крупного сравнительного исследования. Рандомизированное исследование препарата Аредиа указывает на редукцию костных проблем по сравнению с плацебо (“skeletal related events”).
►1997 Вероятность того, что вирусы могут быть вовлечены в «запуск» миеломы. Герпесвирус человека (HHV-8) был найден в дендритических клетках костного мозга. РНК, обнаруженная в крови, имела специфичность к вирусу SV-40, вызывающему
рак у обезьян.
►1998 Продолжающееся исследование роли высокодозной химиотерапии с последующей аутологичной или аллогенной трансплантацией. Реальная польза и характер популяции пациентов, демонстрирующей максимальную пользу от подобной терапии, остаются неясными.
►1999 Показано, что талидамид является эффективным антимиеломным средством при лечении больных с рецидивирующей/ рефрактерной формой заболевания. «Миниаллогенная» трансплантация предложена как менее токсичный подход для достижения эффекта «трансплантат-против-миеломы». Рандомизированное французское исследование показало, что двойная аутологичная трансплантация не имеет дополнительных
преимуществ по сравнению с одиночной. Более долговременное наблюдение показало, что лечение Аредией более двух лет является целесообразным.
►Holmium предложил «фокусное скелетное облучение» в качестве попытки улучшить качество полной ремиссии при аутологичной трансплантации.
►2000 Впервые было представлено несколько новых многообещающих подходов к лечению миеломы. Новые способы терапии миеломы, проходящие клинические испытания, включают аналоги талидамида (“SELCIDS” и “IMIDS”), аналоги адриамицина длительного действия (например, Doxil), триоксид арсения (АТО), агенты антиангиогенеза (например, VEGF , ингибитор тирозин киназы), агенты, блокирующие клеточную адгезию, бетатин и ингибиторы протеазом.
Из всего разнообразия схем химиотерапии, предложенных с 1958 года, ни одна не показала явных преимуществ по сравнению с МР (мелфалан-преднизолон). Комбинация мелфаланпреднизолон обеспечивает объективный ответ у 50-60% больных и имеет клиническую пользу еще у 15-20%.
С 1962 года было опробировано много других схем полихимиотерапии, что, однако, не привело к существенному улучшению результатов лечения. МР, по-прежнему, остается «золотым стандартом» терапии. Хотя высокодозная терапия (например, высокие дозы мелфалана, в/венно) может приводить к более высокому уровню ремиссий у симптоматических больных с далеко зашедшей активной формой болезни (полная ремиссия у 50% больных), еще предстоит определить ее влияние на показатели выживаемости.
Эпидемиология.
ММсоставляет 1% от всех онкологических заболеваний и 10-15% от всех гемобластозов.
Частота ММ в странах Европы и в России колеблется от 3 до 5 на 100000 населения в год, в Америке 3-4 для белых и более 10 – для выходцев из Африки, в странах Азии 2 на 100000 населения.
Наибольшая частота заболевания приходится на возраст 50-70 лет, только 3% - моложе 40 лет. Мужчины и женщины болеют примерно с одинаковой частотой.
Причины развития ММ остаются неясными.
Обсуждается ряд факторов:
-генетическая предрасположенность, связанная скорее всего с дефектами Т-клеточной супрессорной функции;
-влияние хронической антигенной стимуляции; -радиационные и химические воздействия; -вирусные повреждения генома.
Патогенетические механизмы ММ:
ММпредставляет опухоль, возникающую на уровне самых ранних предшественников В-лимфоцитов. Моноклональный пул потомков первично трансформированной клетки сохраняет способность к дифференцировке до конечного этапа – плазматических клеток, секретирующих иммуноглобулины.
Опухолевая трансформация В-лимфоцитов при ММ происходит в терминальном центре периферических лимфоидных органов после соматических гипермутаций реаранжированных гeнов иммуноглобулинов и изотипического переключения синтеза антител. В дальнейшем плазмобласты
иклетки памяти, претерпевшие опухолевую трансформацию, как и
нормальные аналогичные клетки, возвращаются в костный мозг, где при взаимодействии с элементами костно-мозгового окружения проходят окончательный этап созревания до плазматических клеток. В костном мозге эти плазматические клетки формируют опухолевый клон, способный к дальнейшей пролиферации и распространению.
Данные кариологических исследований свидетельствуют о геномной нестабильности, проявляющейся количественными и структурными изменениями хромосом. Наиболее характерными количественными аномалиями кариотипа при ММ являются моносомия хромосомы 13 и трисомия хромосом 3, 5, 7, 9, 11, 15 и 19.
Все генетические события, определяющие возникновение этой опухоли и ее прогрессирование, проходят несколько этапов, которые условно можно разделить на две большие группы — ранние и поздние.
Кранним, или первичным, событиям онкогенеза можно отнести транслокации с вовлечением локуса 14q32 (трансформация во множественную миелому связана с такими дополнительными хромосомными аномалиями, как делеция длинного плеча хромосомы 13). Дальнейшее распространение опухолевого клона обеспечивается микроокружением костного мозга посредством самоподдерживающего механизма взаимодействия миеломных клеток и костно-мозговых стромальных клеток. На этой фазе болезни миеломные клетки зависимы от ростовых факторов и остаются в костном мозге.
Кпоздним генетическим и молекулярным событиям относятся хромосомные аберрации с вовлечением 8q24 (локус гена c-MYC), мутации в протоонкогенах N- и K-RAS, а также мутация ТР53. Эти изменения приводят
кнезависимой от стромы костного мозга пролиферации плазматических клеток с последующим переходом в терминальную фазу болезни и развитием экстрамедуллярных проявлений.
Важную роль в процессе роста опухоли играют цитокины,
секретируемые миеломными клетками и стромальными элементами костного мозга: ИЛ-6, ИЛ-8, ФНО-α, ИНФ-γ, ИЛ-4.
Изучается роль синдекана – 1 (CD138) в патогенезе ММ. При ММ выявляется мутация гена-супрессора опухолевого роста р53. Большое значение придается опухолевому ангиогенезу. Миеломные клетки синтезируют факторы роста эндотелия сосудов (VEGF – vascular endothelial growh factor) и металлопротеиназы (МР), которые взаимодействуя с рецепторами на клетках стромы, стимулируют секрецию ИЛ-6 и ФНО-α.
Характерными чертами ММ является поражение костного мозга (диффузное, диффузно-очаговое, реже очаговое), сопровождающееся костнодеструктивными изменениями (остеопороз, остеолиз) и развитие моноклональной иммуноглобулинопатии (сывороточный М-компонент и/или белок Бенс-Джонса (BJ) в моче). Синдром моноклональной иммуноглобулинопатии – парапротеинемия.
Клиническая симптоматика ММ определяется, с одной стороны,
поражением костного мозга и костей (миелодепрессия, боли, патологические
переломы, костные опухоли, гиперкальциемия), с другой – наличием моноклональной Ig-патии (нефропатия, амилоидоз, полинейропатия, синдром повышенной вязкости, нарушения гемостаза) и вторичным гуморальным иммунодефицитом за счет снижения уровней нормальных Ig (рецидивирующие бактериальные инфекции, синдром недостаточности антител). Висцеральные поражения редки.
Клинико-анатомическая классификация основана на данных рентгенологического исследования скелета и морфологическом анализе пунктатов и трепанатов костей, данных МРТ и КТ. Выделяют диффузноочаговую форму (около 60% наблюдений), диффузную (24%), множественноочаговую (15%), и редкие формы (склерозирующая (<1%) , преимущественно висцеральная (0,5%)).
1)Солитарная миелома (плазмацитома)- есть один очаг миеломной опухоли ( может быть связан с костью или мягкими тканями);
2)Множественно-очаговая- имеется множество костных деструкций на рентгенограмме, нет тотального поражения КМ опухолевыми плазматическими клетками;
3)Диффузно-очаговая (узловая) форманаличие множества костных деструкций и тотальное поражение КМ опухолевыми плазматическими клетками;
4)Диффузнаятотальное поражение КМ опухолевыми плазматическими клетками, но отсутствие костных деструкций. Проявляется тяжёлым анемическим синдромом.
Международная классификация по Салмену и Дьюри. Стадии ММ.
Стадия |
|
Критерии |
Опухолевая |
|
|
|
|
|
масса кг/м2(1012клеток~1кг |
|
|
|
|
опухолевой массы) |
I |
Совокупность следующих признаков: |
До 0,6 (низкая) |
||
|
- Уровень Hb не ниже 100 г/л |
|
||
|
- Нормальный уровень Ca сыворотки |
|
||
|
- Отсутствие остеолиза или солитарный |
|
||
|
костный очаг |
|
|
|
|
-Низкий уровень М-компонента |
|
||
|
↑ Ig J до 50 г/л |
|
|
|
|
↑ Ig A до 30 г/л |
|
|
|
|
- Белок BJ в моче < 4 г/сут. |
|
||
II |
Показатели средние между I и II стадией |
0,6 – 1,2 (средняя) |
||
III |
Один или более последующих признаков: |
Более 1,2 (высокая) |
||
|
- Уровень Hb < 85 г/л |
|
|
|
|
- Уровень Ca сыворотки выше нормы |
|
||
|
- |
Выраженный |
остеодеструктивный |
|
|
процесс (более 3-х костных деструкций) |
|
||
|
-Высокий уровень М-компонента: |
|
||
|
IgG > 70 г/л |
|
|
|
|
IgA > 50 г/л |
|
|
|

Белок BJ в моче > 12 г/сут.
Примечание. Дополнительные стадии А и Б. А – нормальная функция почек
(нормальный уровень креатинина в крови). Б – функция почек нарушена (повышенный уровень креатинина в крови) . Hb – гемоглобин, белок BJ – белок Бенс Джонса.
Практически важным является определение фаз заболевания: хронической (развернутой) или острой (терминальной). Терминальная стадия характеризуется рефрактерностью к ранее эффективной терапии (вторичная резистентность), нарастающей миелодепрессией, прорастанием опухоли в мягкие ткани, внекостномозговыми метастазами, развитием плазмоклеточной лейкемии, иногда периферическим эритрокариоцитозом или миелией.
По степени «агрессивности» ММ (анамнез больных, динамическое наблюдение) различают:
-«тлеющую» ММ без признаков прогрессирования в течение многих месяцев (лет);
-медленно прогрессирующую; -быстро прогрессирующую;
-«агрессивную», в т.ч. ММ-саркому и острый плазмобластный лейкоз.
Диагноз ММ основывается на 3 основных критериях (О.М. Ветякова,
Е.А. Демина, 2007):
1.Плазмоклеточная инфильтрация костного мозга – плазмоцитов более 10% в стернальном пунктате ( в N- не более 1%) (рис. 1);
Рис 1. Пунктат миеломной опухоли (по М. Г. Абрамову, 1985).
2. Моноклональная Ig-патия (сывороточный миеломный компонент и/или белок Бенс-Джонса в моче).
3. Выявление нарушений функций органов или систем, связанных с миеломой (одного или более):
Са (calcium) – гиперкальциемия (содержание кальция в сыворотке крови на 0,25 ммоль/л выше верхней границы нормы или более 2,75 ммоль/л),
R (renal insufficiency) – почечная недостаточность (уровень креатинина в сыворотке крови выше 173 мкмоль/л),
А (anaemia) – анемия (содержание гемоглобина на 2 г/дл ниже нижней границы нормы или менее 10 г/дл),
В (bone lesions) – очаги лизиса в костях или остеопороз.
Диагноз множественной миеломы устанавливают только при наличии не менее 2х из этих трех основных критериев, присутствие первого критерия обязательно.
Клиническая картина.
1. Костно-мозговой синдром. Пролиферирующие в костном мозге миеломные клетки приводят к разрушению костного вещества. В первую очередь деструктивные процессы развиваются в плоских костях и позвоночнике, иногда - в проксимальных отделах трубчатых костей (плечо, бедро); дистальные отделы конечностей и кости лицевого черепа поражаются редко.
Рентгенологические находки зависят от морфологического варианта плазмоцитомы, они убедительны при множественно-очаговой и диффузноочаговой формах и нередко отсутствуют при диффузном поражении костного мозга. Иногда миелому сопровождает развитие очагового или диффузного остеопороза.
В основе поражения костей лежит усиление резорбции костной ткани, связанные с инфильтрацией миеломных клеток, активности остеокластов и нарушением ремоделирования кости. Боли в костях отмечаются у 70% больных. Чаще всего они локализуются по ходу поражения позвонков в пояснично-крестцовой области и грудной клетке, вначале имеют мигрирующий характер, возникая только при перемене положения тела. В поздних стадиях оссалгии становятся нестерпимыми, вынуждая больных лежать неподвижно. Поражение скелета сопровождается костными деформациями. Классический симптом – спонтанные переломы. Боль,
опухоли, переломы – классическая триада Калера – типична для больных ММ в поздних стадиях и при агрессивных формах болезни. У 10% больных развивается синдром сдавления спинного мозга с нижним парапарезом и параплегией. Рентгенологически – генерализованный остеопороз, единичные или множественные
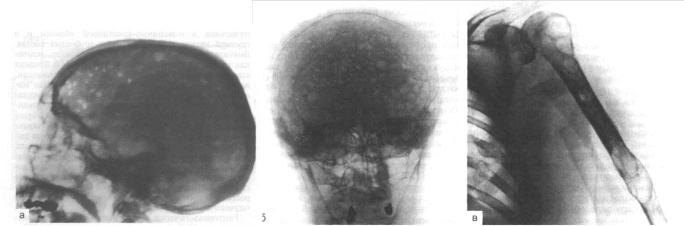
очаги остеолиза, патологические переломы, иногда миелому сопровождает развитие очагового или диффузного остеосклероза. Наиболее характерны поражения в костях черепа: отдельные дефекты выглядят как бы изъеденными молью или выбитыми пробойником. (рис. 2а,б,в).
Рис. 2(а,б,в). Поражение черепа, ребер и плечевой кости у больного ММ (по М.А. Волковой, О.М. Вотяковой, Е.А. Деминой, 2007).
Гиперкальциемия в сыворотке крови наблюдается у 10-30% больных с
ММи обусловлена усиленной резорбцией костей.
Вотличие от большинства гемобластозов, ММ поражает костный мозг в виде дискретных очагов, между которыми сохраняются участки нормального кроветворения или зоны с минимальным количеством опухолевых клеток.
Т.о основными проявлениями костно-мозгового синдрома являются: - значительная гиперплазия в КМ опухолевых плазматических клеток
- остеопороз, с последующим развитием очаговых деструкций (с-м пробойника, ребра «изъеденные молью», рыбьи позвонки)
- на конечных этапахтриада Калера ( боль, пат.опухоль, пат.перелом).
2. Синдром белковой патологии.
►Миеломная нефропатия (парапротеинемический нефроз) – наиболее частое и серьезное проявление парапротеинемии. Почечная недостаточность занимает одно из первых мест среди причин смерти больных. Клиника миеломной нефропатии складывается из упорной протеинурии и постепенно развивающейся почечной недостаточности. При этом отсутствуют классические признаки нефротического синдрома: отеки, гипопротеинемия, гиперхолестеринемия, нет симптомов сосудистых почечных поражений – гипертонии, ретинопатии.
В различных стрессовых ситуациях (переломы костей, инфекции, лекарственная непереносимость и пр.) у больных миеломной болезнью наступает острая почечная недостаточность или резко декомпенсируется имевшийся до этого почечный процесс, нередко с развитием олигоили анурии и быстрым нарастанием азотемии.
►Амилоидоз выявляется в среднем у 1015% больных миеломной болезнью. Основным компонентом амилоидных фибрилл являются моноклональные легкие цепи или их вариабельные участки (AL-амилоидоз).
В первую очередь и, главным образом, поражаются органы, богатые коллагеном: адвентиция сосудов, мышцы (сердце, язык), дерма, сухожилия и суставы. Печень, селезенка, почки обычно не страдают или амилоидные отложения в них бывают очень скромными. Следует помнить, что без других причин нарастающая глухость сердечных тонов, упорная тахикардия, снижение вольтажа ЭКГ, сердечная недостаточность, макроглоссия, разнообразные дерматозы, упорные ревматоидные боли в суставах с их деформацией, синдром карпального канала, диспепсические расстройства, упорный геморрагический синдром, дистрофия роговицы при миеломной
болезни могут быть следствием параамилоидных отложений в
соответствующих органах. В ряде случаев параамилоид образует массивные локальные псевдоопухолевые узлы по ходу желудочно-кишечного тракта, иногда преимущественно откладывается в слюнных и щитовидной железах, отдельных группах лимфатических узлов.
► Синдром NAMIDD – неамилоидное отложение в тканях моноклональных L- и Н-цепей или целых молекул. В отличие от амилоидоза,
при NAMIDD отложения имеют аморфный нефибриллярный состав и не выявляются гистологическими методами, принятыми для диагностики амилоидоза. В их составе при помощи моноспецифических антииммуноглобулиновых антисывороток выявляются в основном L-цепи - типа. Поражаются, главным образом, почки (нефротический синдром), сердце (нарушение ритма и проводимости, снижение сократительной способности миокарда), мышцы (рабдомиолиз), ЖКТ (нарушение всасывания, диарея), кожа, суставы, слюнные железы. Описаны отложения в легких (диспноэ), верхних дыхательных путях ,печени (гепатомегалия, повышение уровня трансаминаз, портальная гипертензия). Окончательный диагноз возможен только путем иммуногистологического исследования биоптатов пораженных органов.
3.Синдром недостаточности антител. Характерным симптомом множественной миеломы является резкое снижение уровня нормальных иммуноглобулинов. По мере прогрессирования заболевания их уровень закономерно снижается (гуморальный иммунодефицит закономерно нарастает) вплоть до полного их исчезновения, что выражается склонностью больных к бактериальным инфекционным осложнениям, особенно с локализацией в носоглотке, дыхательных и мочевыводящих путях.
4.Синдром повышенной вязкости (↑общего белка до 150 и более г\л)
характеризуется кровоточивостью слизистых оболочек, геморрагической ретинопатией, расширением вен сетчатки (fundus paraproteinaemicus), нарушениями периферического кровотока, парестезиями, синдромом Рейно,
втяжелых случаях изъязвлениями и даже гангреной дистальных отделов конечностей. Нарушения микроциркуляции в сосудах головного мозга могут служить причиной парапротеинемической комы. При криоглобулинемии эти симптомы резко выражены после охлаждения.
5.Синдром висцеральных поражений. У 5-13% больных констатируют гепато- (или) спленомегалию. Приблизительно у половины пациентов увеличение органов связано со специфической миеломноклеточной пролиферацией, у остальных цитологический состав пунктатов и отпечатков селезенки выявляет смешанную миеломномиелоидную или чисто миелоидную трехростковую пролиферацию. Характерным гематологическим симптомом у этих больных является миелемия (резкий левый сдвиг в лейкоцитарной формуле), нередко с эритрокариоцитозом. Поражение лимфатических узлов встречается в 0,5% случаев. Специфическое поражение желудка может проявляться в виде инфильтративного процесса, язвы или плазмоцитомы. Симптоматическая периферическая нейропатия наблюдается у 5-15% больных: нарушения