

Федеральное государственное автономное образовательное учреждение
высшего образования «СИБИРСКИЙ ФЕДЕРАЛЬНЫЙ УНИВЕРСИТЕТ»
Институт фундаментальной биологии и биотехнологии
Базовая кафедра биотехнологии
Митохондриальные болезни – большая группа наследственных заболеваний, обусловленных нарушением структуры и биохимических процессов в митохондриях.

Особенности строения и функционирования мтДНК по
сравнению с ядерным геномом.
отсутствие интронов, вследствие чего наблюдается высокая плотность и значительное уменьшение промежутков между генами по сравнению с ядерной ДНК;
большинство митохондриальных мРНК не содержат 5’- и 3’-нетранслируемые последовательности;
репликация мтДНК асинхронный
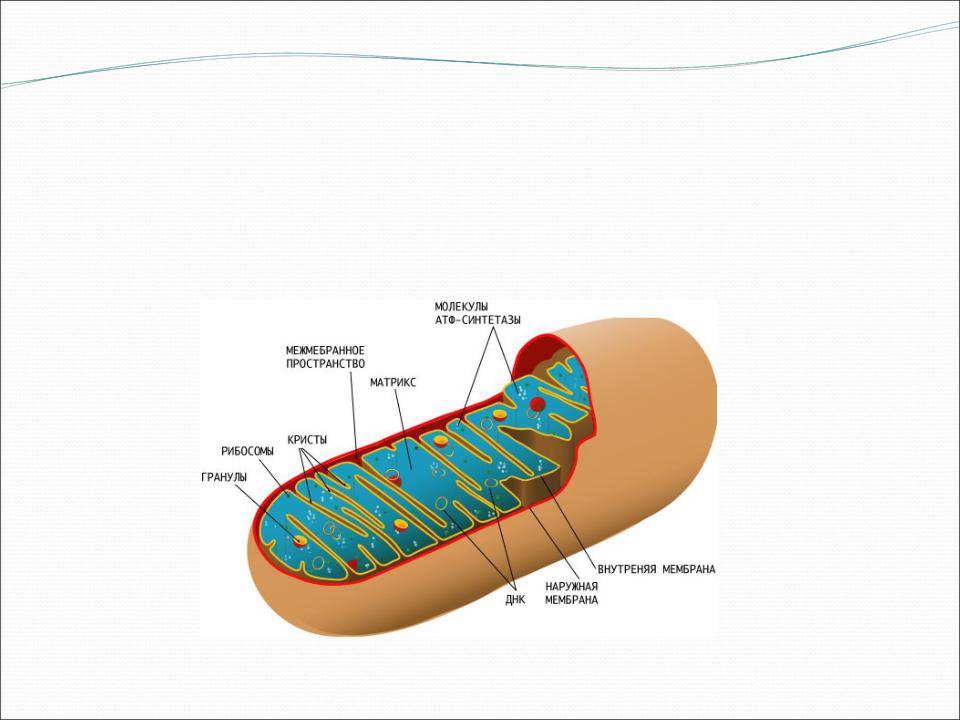
МтДНК содержит:
•гены, которые кодируют 13 полипептидов, входящих в пять комплексов дыхательных цепей митохондрий;
•22 гена транспортной РНК;
•2 гена рибосомной РНК.
Остальные 70 белков, участвующих в процессах окислительного фосфорилирования, находятся под контролем ядерных генов.

Классификация
митохондриальных болезней на основе этиологии
1) заболевания, обусловленные мутациями в генах ядерной ДНК;
2) заболевания, связанные с мутациями в митохондриальном геноме;
3) заболевания, вызванные нарушением межгеномных сигнальных эффектов.
Острая энцефалопатия с отёком мозга и жировой инфильтрацией органов (преимущественно, печени), возникает у ранее здоровых новорождённых, детей и подростков (чаще в возрасте 4–12 лет), часто связан с предшествующей вирусной инфекцией (например, ветряная оспа или грипп А) и приёмом препаратов, содержащих ацетилсалициловую кислоту.
Органы мишени - ЦНС, печень, мышцы
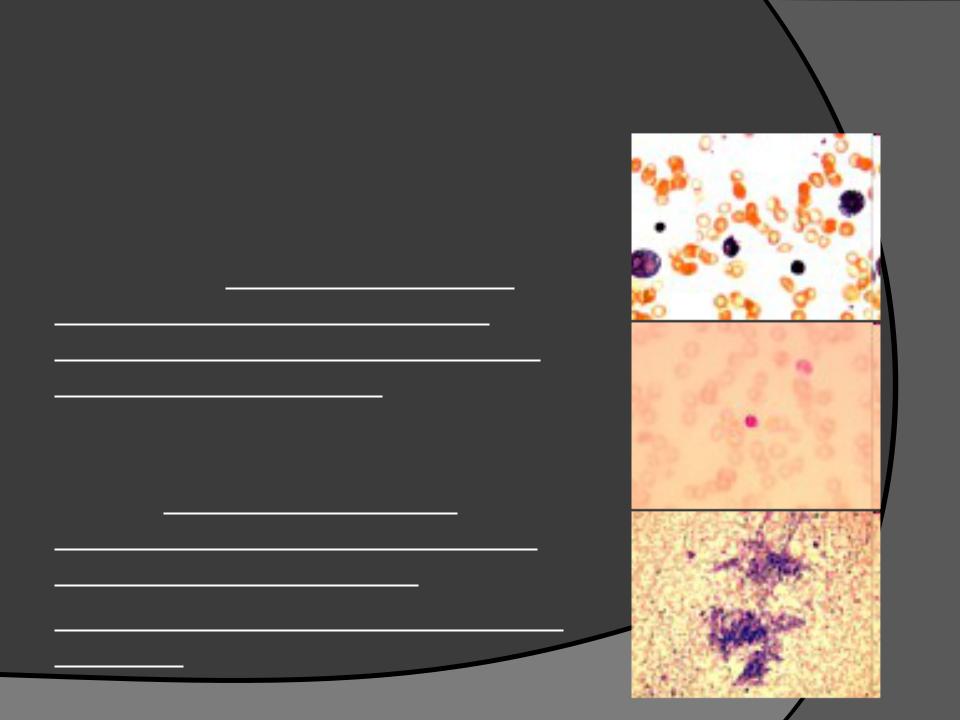
Синдром Пирсона ( Pearson syndrome )
Был описан Pearson в 1979 г. Характерными признаками считают:
Упорную сидеробластную анемию с вакуолизацией эритроидных и миелоидных предшественников.
Пациенты могут иметь зависимую от переливаний крови макроцитарную
анемию с нейтропенией и тромбоцитопенией.
Дисфункцию поджелудочной железы

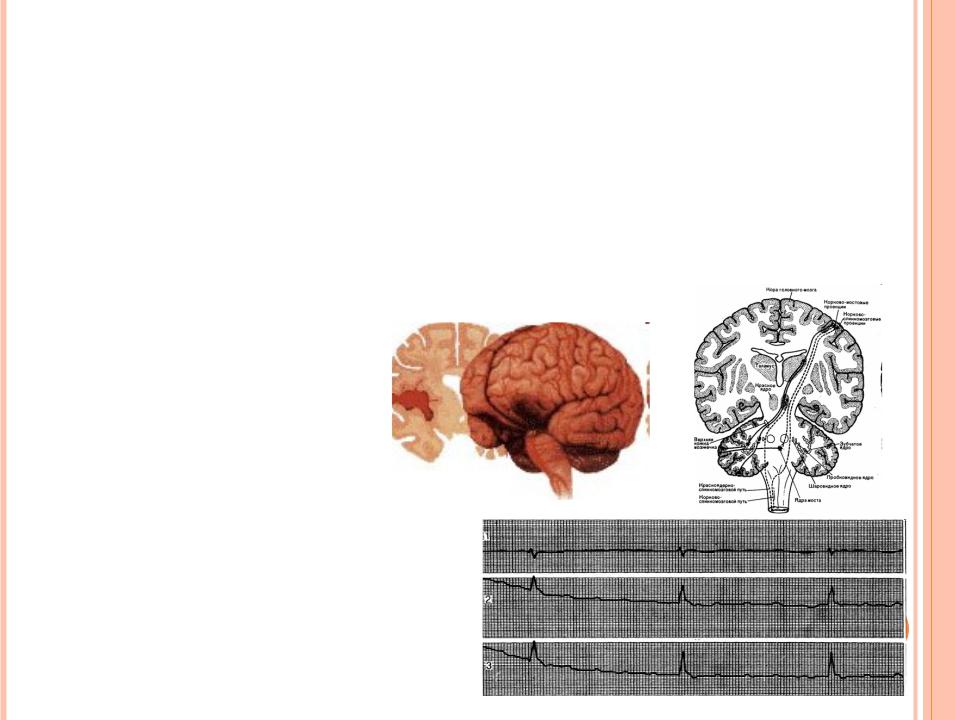
СИНДРОМ КЕРНСАСЕЙРА
Впервые описан Кернсом в 1946 году. Более детальное изучение этого заболевания принадлежит Сейру в 1956 году. Характерный клиническим симптомокомплекс:
прогрессирующая
наружная
офтальмоплегия,
пигментная ретинопатия,
КМП с нарушением проводящей системы и развитием полного атриовентрикулярного блока.
Пониманию природы синдрома способствовали молекулярногенетическиеисследования и
обнаружение мутаций митохондриальной ДНК

Отмечается задержка физического и полового развития.
Изменения со стороны кожи проявляются ихтиозом с очагами гиперпигментации.
Нарушения со стороны опорнодвигательного аппарата характеризуются вальгусной девиацией голеней и высоким сводом стопы.
Патология со стороны глаз наружная офтальмоплегия с птозом различной степени выраженности, пигментный ретинит или пигментная дегенерация сетчатки. Птоз — наиболее типичный признак.