

Posobie_farmakologia_chast_1
.pdf151
ние. При этом сам пациент не осознает своего временного отключения и продолжает выполнять ту работу (беседу, письмо, еду и т.п.) за которой его застиг припадок.
Миоклонус-эпилепсия является одной из самых неблагоприятных форм эпилепсии, она плохо поддается медикаментозной коррекции и быстро приводит к формированию дефекта личности. Чаще всего эта форма эпилепсии начинается после органического поражения головного мозга – энцефалита, отравления нейротропными ядами, уремии или гипоксии. Миоклонус характеризуется внезапным возникновением частых подергиваний групп мышц при полном сохранении сознания, обычно миоклонический припадок может многократно повторяться с интервалами в несколько минут.
Простые парциальные судороги характеризуются возникновением приступа судорог в группе мышц, которая соответствует локализации эпилептогенного очага (судороги одной руки, судороги пальца и т.п.). Часто эпизоду судорог предшествует аура. Эти припадки протекают при сохранении сознания.
Сложные парциальные припадки характеризуются отсутствием типичной судорожной картины как таковой. У пациента наблюдается сумеречное расстройство сознания с возникновением галлюцинаций, нарушением мышления, возникновением психомоторного автоматизма (выполнение сложных целенаправленных движений и поступков без контроля со стороны сознания). Как правило, после выхода из припадка у человека наблюдается антероградная амнезия на события, которые с ним происходили во время припадка.
Отдельно выделяют эпилептический статус – особое состояние, при котором судорожные припадки следуют друг за другом, при этом завершение одного припадка происходит в тот момент, когда начинается следующий. Основным критерием эпилептического статуса является отсутствие даже кратковременного периода возвращения пациента в нормальное состояние между припадками.
Сравнительная характеристика различных типов припадков показана в таблице 1.
Таблица 1. Сравнительная характеристика судорожных припадков при эпилепсии.
|
Припадок |
|
|
Аура |
|
|
Нарушение сознания |
|
|
Судороги |
|
|
Амнезия |
|
|
|
|
|
|
|
|
|
|
|
|||||
Grand mal |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Абсанс (petit mal) |
|
|
▬ |
|
|
|
|
|
▬ |
|
|
|
|
|
Миоклонус |
|
|
▬ |
|
|
▬ |
|
|
|
|
|
▬ |
|
|
Простые парциальные |
|
|
|
|
|
▬ |
|
|
|
|
|
▬ |
|
|
Сложные парциальные |
|
|
|
|
|
|
|
|
▬ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Классификация противоэпилептических средств. До настоящего времени нет обще-
принятой классификации данной группы лекарственных средств. Наиболее удобной является химико-фармакологическая классификация, которая учитывает строение и механизм действия противоэпилептических средств. Согласно этой классификации можно выделить 4 группы противоэпилептических средств.
A.Средства, угнетающие активность Na+-каналов мембран нейронов:
1.производные гидантоина – фенитоин;
2.производные иминостильбена – карбамазепин.
B.Средства, угнетающие активность Ca2+-каналов T- и L-типов:
1.производные сукцинимидов – этосуксимид.
C.Средства, повышающие активность ГАМК-ергической системы:
1.производные барбитуровой кислоты – фенобарбитал;
2.производные жирных кислот – натрия и магния вальпроат;
3.синтетические аналоги ГАМК – габапентин;
4.производные бензодиазепинов – клоназепам, диазепам.
D.Средства, угнетающие активность глутаматергической системы:
1.производные фенилтриазинов – ламотриджин.
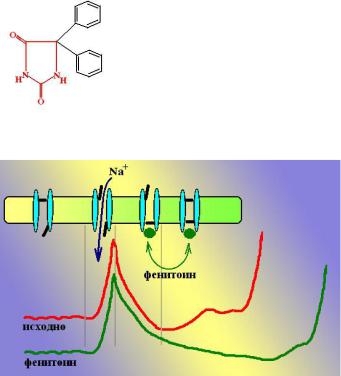
152
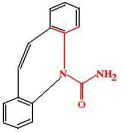
Фенитоин (Phenytoin, Diphenin, Dylantine). Является производным гидантоина. Был синтезирован в 1908 г как снотворное средство, аналог барбитуратов. Однако, он не нашел практического применения, так как практически не обладал седативным и снотворным эффектом. В 1938 г в процессе скриннинга противоэпилептических средств была обнаружена его противосудорожная активность.
Механизм действия: Показано, что фенитоин связывается с инактивированными натриевыми каналами и продлевает это состояние, замедляя реактивацию канала. Таким образом, поступление Na+ внутрь клетки и генерация потенциала действия затрудняется. Поскольку активность нейронов в эпилептогенном очаге значительно выше, чем в нормальной ткани и ПД генерируются там значительно чаще, количество инактивированных натриевых каналов в таких очагах значительно больше, чем в обычных нейронах,
поэтому фенитоин подавляет преимущественно активность нейронов эпилептогенного очага. Кроме того, установлено, что фенитоин обладает также рядом других механизмов действия:
фенитоин нарушает ток ионов кальция в клетку по кальциевым каналам, это нарушает процесс деполяризации и генерации длительных возбуждающих потенциалов, необходимых для образования гиперсинхронных пачек нервных импульсов.
В высоких концентрациях фенитоин подавляет процесс выделения в синапс норадреналина и серотонина и нарушает обратный нейрональный захват дофамина. В конечном итоге, это приводит к затруднению передачи в моноаминергических синапсах ЦНС.
ФК: после перорального приема фенитоин хорошо и полностью всасывается, максимальная концентрация фенитоина в плазме крови возникает через 4-12 ч. Однако, следует помнить, что скорость и полнота абсорбции фенитоина отличаются у таблеток, произведенных разными фармацевтическими компаниями, поэтому самостоятельная замена одного препарата фенитоина на другой (даже аналогичной дозировки) без контроля врача недопустима. Возможно внутривенное введение натриевой соли (phenytoin sodium) или фосфорного эфира (fosphenytoin) фенитоина, при этом следует помнить, что нельзя растворять фенитоин в растворах глюкозы, так как возможно его окисление. Внутримышечное введение фенитоина недопустимо, поскольку в мышцах фенитоин образует плохорастворимые преципитатыи всасывается медленно и неполно (при внутримышечном введении биодоступность фенитоина меньше, чем при его пероральном введении).
В крови фенитоин на 90% связан с белками крови и уровень свободной фракции составляет около 10%. В том случае, если человек одновременно принимает другие средства, которые также способны интенсивно связываться с белком (сульфаниламидные препараты, сахароснижающие средства, нестероидные противовоспалительные средства, оральные антикоагулянты и др.), происходит конкуренция за белки крови и часть фенитоина может переходить в свободное состояние. Вытеснение даже 10% фенитоина из связи с белком приводит к тому, что уровень его свободной фракции увеличивается в 2 раза (возрастание с 10% до 20%) и соответственно в 2 раза возрастает риск нежелательных эффектов и интоксикации.
Для фенитоина характерна нелинейная кинетика – в малых дозах скорость его элиминации пропорциональна введенной дозе и концентрация фенитоина постепенно стабилизируется на некотором стационарном уровне (кинетика первого порядка), но как только величина дозы фенитоина превысит метаболические возможности ферментов печени, кинетика фенитоина переходит в нулевой порядок, т.е. скорость элиминации остается постоянной и не

153
зависит от дозы. Поэтому даже при незначительном увеличении дозы возможен резкий скачок концентрации фенитоина, который приводит к развитию отравления.
Биотрансформация фенитоина происходит в печени путем глукуронидации и гидроксилирования (при участии цитохромов Р450). В процессе лечения фенитоином происходит индукция (повышение активности) этих ферментов, что может привести к увеличению разрушения как самого фенитоина, так и других лекарств, принимаемых совместно с ним.
ФЭ и показания к применению:
1.Фенитоин оказывает противосудорожное действие при парциальных эпилептических припадках, grand mal; при этом в терапевтических дозах он практически не влияет на ког- нитивно-мнестические процессы, т.к. не изменяет функции нормальных нейронов.
2.Фенитоин способен купировать судороги при эпилептическом статусе, при этом обычно используют внутривенное введение натриевой соли или фосфорного эфира фенитоина, реже прибегают к ректальному введению при помощи микроклизм.
3.Фенитоин устраняет боль при невралгии тройничного нерва (заболевание, при котором даже минимальное раздражение нерва – например дуновение ветра в лицо, вызывает тяжелый приступ болей). Данный эффект нельзя назвать анальгетическим, поскольку проявляется он только при невралгии и не возникает при любых других видах болей.
4.Фенитоин оказывает противоаримическое действие, которое связано с его способностью блокировать натриевые каналы мембран кардиомиоцитов (мембраностабилизирующее действие). В отличие от всех других противоаритмических средств, фенитоин не оказывает никакого влияния на проводящую систему миокарда, поэтому он особенно эффективен при аритмиях, вызванных передозировкой сердечных гликозидов (при этих аритмиях
повышается возбудимость только рабочего миокарда, а возбудимость проводящей системы не изменяется или даже понижается).
РД: Ввиду того, что кинетика фенитоина нелинейна, подбор и изменение дозы фенитоина следует выполнять крайне осторожно. Вначале назначают нагрузочную дозу фенитоина – с интервалом в 2 часа пациент должен принять вначале 3 таблетки, затем еще раз 3 таблетки и наконец 2 таблетки фенитоина. Таким образом, всего за 4 часа он принимает 8 таблеток фенитоина.
После этого пациента переводят на поддерживающую дозу фенитоина. Он принимает по 3 таблетки фенитоина 1 раз в день (как правило утром). При необходимости дозу фенитоина повышают на ¼ таблетки каждую неделю. Максимальная возможная поддерживающая доза 4 таблетки в день. Детям фенитоин назначают в дозе 5 мг/кг веса в день.
При лечении фенитоином следует стремиться поддерживать его концентрацию на терапевтическом уровне. О концентрации фенитоина в крови судят либо по лабораторным данным мониторирования концентрации, либо косвенно, по сопутствующим каждому уровню клиническим симптомам:
Таблица 2. Клинические симптомы при различных уровнях фенитоина в крови.
|
Концентрация фенитоина в крови |
|
|
Уровень, симптоматика |
|
|
|
|
|
|
|||
<5 мкг/мл (<20 мкмоль/л) |
|
|
Субклинический уровень |
|||
5-20 мкг/мл (20-80 мкмоль/л) |
|
|
Терапевтический уровень (горизонтальный нистагм) |
|||
20-30 |
мкг/мл (80-120 мкмоль/л) |
|
|
Субтоксический уровень (диплопия, нарушения дви- |
||
|
|
|
|
|
жения глаз) |
|
30-40 |
мкг/мл (120-160 мкмоль/л) |
|
|
Токсический уровень (атаксия) |
||
>40 мкг/мл (>160 мкмоль/л) |
|
|
Сублетальный уровень (нарушение психики, галлюци- |
|||
|
|
|
|
|
нации, летаргия) |
|
При использовании фенитоина в качестве противоаритмического средства его вводят внутривенно медленно в дозе 4 мг/кг веса под контролем ЭКГ.
НЭ: Различают нежелательные эффекты фенитоина, которые проявляются при низком и при высоком уровне лекарства.
При низком уровне лекарства:
154
♦гипертрофия десен (возникает в 20% случаев) – чаще появляется у молодых лиц, выраженность ее может быть уменьшена при соблюдении гигиены полости рта;
♦гирсутизм, огрубение черт лица, акне;
♦реакции гиперчувсвительности – сыпи, лимфаденопатия, волчаночный синдром;
♦гематотоксические реакции – нейтропения, мегалобластная анемия (обусловлена увеличением выведения и снижением абсорбции витамина Вс);
♦остеопороз и остеомаляция (снижение чувствительности периферических тканей к витамину D, нарушение обмена кальция);
♦снижение выделения инсулина поджелудочной железой (гипергликемические состояния);
♦прием фенитоина во время беременности приводит к развитию гидантоинового синдрома плода – сочетания микроцефалии с расщеплением губы, незаращенным небом, гипоплазией ногтей и фаланг пальцев. Этот эффект обусловлен образованием ареноуксусного метаболита фенитоина;
♦толерантность (привыкание) к фенитоину и снижение эффекта других лекарственных средств на фоне фенитоина, что обусловлено индукцией микросомальных ферментов при его применении (особого внимания требуют пациентки, принимающие оральные контрацептивы, т.к. у них необходимо увеличить дозу контрацептивов);
♦синдром рикошета – возникает при внезапном прекращении применения фенитоина, характеризуется резким возрастанием числа судорожных эпизодов у пациента.
При высоком уровне лекарства:
♦нейротоксические реакции – атаксия, головокружение, диплопия. Нистагм, сонливость, нарушение поведения, галлюцинации, судороги;
♦тошнота и рвота (эти эффекты можно уменьшить, если принимать высокие дозы фенитоина во время еды);
♦при внутривенных инъекциях возможно локальное повреждение сосудов с развитием отека и обесцвечивания кожи по ходу вены, снижение артериального давления, развитие аритмии.
ФВ: табл. 0,117
Карбамазепин (Carbamazepin, Tegretol, Finlepsin). По химическому строению весьма близок к имипрамину. Был введен в клиническую практику в 1960 г, как альтернатива имипрамину, однако, в связи с низкой антидепрессивной активностью он длительно применялся как средство для лечения невралгии тройничного
нерва. С 1974 г. используется как противоэпилептическое средство.
МД: Трехмерная конфигурация карбамазепина весьма близка к конформации фенитоина, поэтому он также способен связываться с инактивированными натриевыми каналами и пролонгировать состояние их инактивации, нарушая поступление ионов натрия в клетку и генерацию ПД.
Относительно недавно было установлено, что карбамазепин является антагонистом NMDA-рецепторов для глютаминовой кислоты. Карбамазепин взаимодействует с активным центром этих рецепторов и препятствует их активации глютаминовой кислотой. Поскольку рецепторы остаются неактивными не происходит открытия сопряженного с ними кальциевого канала и нарушается поступление ионов кальция в клетку. Поступление кальция в пресинаптическое окончание необходимо для выделения медиаторов, а поступление этих ионов в постсинаптическое окончание – для генерации длительных возбуждающих потенциалов. Оба этих процесса под влиянием карбамазепина нарушаются.
Кроме того, поступление избытка ионов кальция в нейрон во время судорожной активности приводит к перегрузке ионами кальция митохондрий и их гибели. Карбамазепин оказывает защитный эффект и уменьшает тем самым дегенеративные процессы в нейронах под влиянием избытка ионов кальция.
ФК: Карбамазепин хорошо всасывается после перорального применения, с белками крови связывается на 60-70%, но практически не вытесняется другими лекарственными

155
средствами из связи с белком и поэтому совместно с другими лекарствами может применяться без дополнительной коррекции дозы.
При регулярном использовании карбамазепин вызывает индукцию микросомальных ферментов печени и усиливает свой собственный метаболизм. Поэтому с течением времени скорость выведения карбамазепина возрастает (вначале его период полуэлиминации составляет около 36 часов, а затем снижается до 20 часов).
В процессе метаболизма карбамазепина образуется 10,11-эпоксикарбамазепин, который обладает собственной противосудорожной активностью, составляющей 30% активности карбамазепина.
ФЭ и показания к применению:
1. Противосудорожный эффект. Карбамазепин является средством выбора при лечении сложных парциальных припадков (психомоторных эквивалентов), кроме того, он эффективен при простых парциальных припадках и grand mal.
2. Карбамазепин устраняет боль при невралгии тройничного нерва. Эффект карбамазепина превосходит эффект фенитоина и является весьма специфичным именно для этого за-
болевания, поэтому иногда карбама-
Схема 2. Механизм действия карбамазепина и барбитуратов зепин применяют с диагностической
целью при боли в области лица неясного генеза (если карбамазепин снимает эту боль, то диагноз невралгии считается подтвержденным).
3. Карбамазепин обладает способностью «стабилизировать» настроение, что используется при маниакальной стадии маниакально-депрессивного психоза как альтернатива солям лития.
РД: Карбамазепин применяют исключительно перорально в дозе 1,0-2,0 г/сут, разделенной на 3-4 приема. Детям карбамазепин назначают в дозе 15-25 мг/кг/сут.
НЭ:
1.Диплопия – для нее характерна строгая периодичность, двоение в глазах возникает в определенное время суток и продолжается около часа. Часто ее можно устранить просто изменив время приема препарата.
2.Атаксия.
3.Сонливость, вялость, головокружение.
4.Отеки и гипонатриемия (особенно выражены у пожилых людей). Полагают, что отеки связаны со способностью карбамазепина усиливать секрецию АДГ.
5.Тошнота, диарея, усугубление судорог.
6.Снижение эффективности комбинированных оральных контрацептивов, вследствие индукции микросомальных ферментов печени.
7.Апластическая анемия и агранулоцитоз – чаще всего эти эффекты возникают у пожилых лиц, которые принимают карбамазепин в связи с невралгией тройничного нерва.
8.Идиосинкразия – протекает в форме эритемы кожи и фотосенсибилизации.
ФВ: табл. 0.1 и 0,2; таблетки ретард по 0,2 и 0,4.
Этосуксимид (Ethosuximide, Suxilep). Создан в 1960 г. в США. Механизм действия: Этосуксимид блокирует низкопороговые кальциевые каналы (T и N-типа), которые обеспечивают спонтанную деполяризацию мембран таламических нейронов с частотой 3 Гц, выступая в роли своеобразного «водителя ритма». В итоге, нарушается генерация ритмических разрядов таламуса, которая запускает приступ абсанса.

156
Показано, что в высоких дозах этосуксимид ингибирует Na+,K+-АТФазу и нарушает тем самым процесс реполяризации мембраны, продлевая состояние сниженной возбудимости нейронов.
В высоких дозах этосуксимид ингибирует активность ГАМК-транаминазы (ГАМК-Т), фермента, который разрушает ГАМК до янтарного полуальдегида, поэтому в синапсах ЦНС возрастает количество ГАМК, которая оказывает тормозящее влияние на нейроны.
ФК: Этосуксимид практически полностью реабсорбируется из ЖКТ, практически не свяхывается с белками крови, поэтому прием других лекарств не оказывает никакого влияния на его уровень в крови. Метаболизм этосуксимида происходит в печени с образованием гироксипроизводного под влиянием ферментов системы цитохрома Р450. Выведение этосуксимида крайне медленное, период полуэлиминации составляет около 40 часов (по некоторым данным до 72 часов), т.е. полное выведение этосуксимида после однократного применения занимает около 2 недель.
ФЭ: Этосуксимид является лекарственным средством с очень узким спектром активности. Он оказывает эффект только при абсансах.
Для достижения терапевтического эффекта требуется прием 750-1500 мг этосуксимида в день. Поскольку период полуэлиминации этосуксимида достаточно продолжительный, его можно было бы применять 1 раз в день, однако этому мешает мощное раздражающее влияние этого лекарства на слизистые оболочки ЖКТ (боли в желудке, тошнота, рвота), поэтому дозу этосуксимида обычно применяют в 2 приема.
НЭ: Наиболее значимым является раздражающее действие этосуксимида на слизистые оболочки ЖКТ (его можно уменьшить, если начинать прием этосуксимида с малых доз и делить суточную дозу на 2 приема). Реже возможны преходящая сонливость, усталость.
ФВ: капс. 250 мг
Фенобарбитал (Phenobarbital, Luminal). Производное барбитуровой кислоты, одно из старейших противоэпилептических средств (для лечения эпилепсии применяется с 1912 г).
Длительное время являлся средством выбора при лечении различных форм эпилепсии. В настоящее время его относят к средствам резерва (альтернативным средствам).
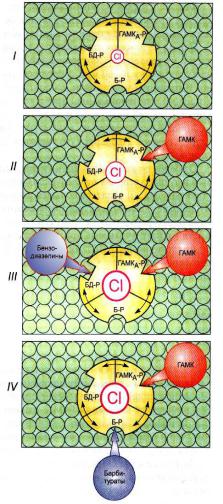
Механизм действия: Фенобарбитал активирует барбитуровый участок алостерического центра ГАМКА-хлоридного ионофорного комплекса, который включает в себя хлидный канал, ГАМКА-рецептор и 2 аллостерических центра – барбитуровый и бензодиазепиновый. Активация барбитурового
центра комплекса приводит к резкому повышению сродства ГАМКА-рецепторов к ГАМК, при этом даже ничтожные количества ГАМК могут активировать рецептор и открыть хлоридный канал, при этом под влиянием фенобарбитала длительность пребывания канала в открытом состоянии также резко увеличивается. Ионы хлора поступая в клетку через открытый канал вызывают гиперполяризацию мембраны нейрона эпилептогенного очага и снижают его возбудимость.
Показано, что фенобарбитал блокирует АМРА-рецепторы для глютаминовой кислоты. АМРА-рецепторы связаны с натриевыми каналами мембраны. Их блокада приводит к тому, что глютамат не способен активировать рецептор и каналы остаются закрытыми. Прекращается поступление ионов натрия в клетку и генерация ПД. Кроме того, поступление избыточных количеств натрия в нейроны при судорожном припадке способствует развитию отека клетки, т.к. по осмотическому градиенту вслед за натрием в клетку поступает вода. Поскольку фенобарбитал снижает поступление натрия в нейрон, он косвенно способствует уменьшению отека нервной ткани.
В чрезвычайно больших количествах (которые не создаются в организме при обычной противосудорожной терапии) фенобарбитал способен блокировать Na+- и Са2+-каналы (L- и N-типов), что также снижает возможности генерации клеткой возбуждающих потенциалов.
ФК: После перорального применения всасывается медленно. Фенобарбитал является слабой кислотой с рКа=7,3 поэтому при рН крови около 50% принятого лекарства находится
157
в ионизированном состоянии и в ЦНС способно проникать только ½ принятой дозы. Во время судорог развивается гипоксия, которая приводит к ацидозу и доля неинонизированного фенобарбитала в крови повышается, а следовательно увеличивается его поступление в ЦНС и усиливается противосудорожный эффект лекарства.
Метаболизм фенобарбитала протекает в печени при участии цитохромов Р450. В процессе метаболизма происходит индукция (увеличение активности) микросомальных ферментов печени и ряда других ферментных систем гепатоцитов (аланиновой синтетаза, -глютамил трансаминазы, глюкуронидазы и др.). Поэтому эффект ряда лекарственных средств на фоне приема фенобарбитала ослабляется (это обусловлено увеличением из разрушения).
Выводится фенобарбитал крайне медленно, период полуэлиминации для него составляет 4-5 дней, поэтому для проведения адекватной противосудорожной терапии достаточно однократного применения фенобарбитала в сутки. Элиминация фенобарбитала осуществляется почками, при этом 50% вве-
денного лекарства выводится в неизмененном виде. ФЭ:
|
|
1. |
Противосудорожный эффект – в настоящее время фе- |
|
|
|
|
нобарбитал является препаратом резерва при лечении |
|
|
|
|
grand mal, миоклонус эпилепсии и простых парциаль- |
|
|
|
|
ных припадков. Он малоэффективен при абсансах и |
|
|
|
|
сложных парциальных припадках. Растворимая форма |
|
|
|
|
фенобарбитала (фенобарбитал натрия) применяется для |
|
|
|
|
купирования эпилептического статуса. |
|
|
|
2. |
Седативный эффект – фенобарбитал повышает актив- |
|
|
|
|
ность ГАМКА-хлоридного комплекса и усиливает про- |
|
|
|
|
цессы торможения в ЦНС. При регулярном приеме у |
|
|
|
|
этому эффекту развивается быстрое привыкание (толе- |
|
|
|
|
рантность), однако следует помнить, что толерантность |
|
|
|
|
никогда не развивается к противосудорожному эффек- |
|
|
|
|
ту фенобарбитала. |
|
|
|
3. |
Снотворный эффект – возникает в дозах, значительно |
|
|
|
|
превышающих противосудорожные. Сон, который вы- |
|
|
|
|
зывает фенобарбитал отличается от физиологического |
|
|
|
|
и после пробуждения человек испытывает чувство раз- |
|
|
|
|
битости и усталости, не ощущает «прилива сил». Фе- |
|
|
|
|
нобарбитал удлиняет время сна и его глубину, но прак- |
|
|
|
|
тически не влияет на процесс засыпания. |
|
|
|
4. |
Для фенобарбитала характерен ряд нейрометаболиче- |
|
|
|
|
ских эффектов – он снижает потребность нейронов в |
|
|
|
|
кислороде (антигипоксический эффект), понижает |
|
|
|
|
внутричерепное давление, способствует перераспреде- |
|
Схема 3. Механизм |
активации |
|
лению крови в зоны ишемии, подавляет процессы ПОЛ |
|
ГАМКА-рецептор хлоридного ионо- |
|
в мембранах нейронов. |
||
форного комплекса. I – |
состояние |
|
||
|
НЭ: |
|||
покоя; II – повышение проводимости |
|
|||
1. |
Седативное и снотворное действие фенобарбитала не- |
|||
канала под влиянием ГАМК; III – уве- |
||||
личение частоты открытия канала |
|
желательно у молодых пациентов, которые ведут ак- |
||
под влиянием ГАМК на фоне бензодиа- |
|
тивный образ жизни. |
||
зепинов (БД-Р – бензодиазепиновый |
2. |
При длительном применении вследствие индукции фер- |
||
аллостерический центр); |
IV – увели- |
|||
чение длительности открытия канала |
|
ментов печени под влиянием фенобарбитала может ос- |
||
|
лабляться эффект других лекарственных средств, которые |
|||
под влиянием ГАМК на фоне барбиту- |
|
|||
ратов (Б-Р – барбитуровый аллосте- |
|
применяются совместно с фенобарбиталом. Иногда, этот |
||
рический центр). |
|
|
эффект фенобарбитала используют с лечебной целью у |
|
По Д.А. Харкевичу, 1999 |
|
пациентов с синдромом Жильбера (недостаточность |
||
|
|
|
||

158
УДФ-глюкуронидазы – фермента печени обеспечивающего перевод непрямого билирубина в прямой, под влиянием фенобарбитала происходит индукция этого фермента).
3.Длительный прием фенобарбитала приводит к формированию барбитуровой наркотической зависимости со снижением интеллекта, памяти, способности к обучению. Прекращение приема фенобарбитала сопровождается развитием абстинентного синдрома с нарушением поведения, резким возбуждением, судорогами и часто завершается летальным исходом.
4.Фенобарбитал ускоряет метаболизм витамина D, приводя к развитию рахитоподобных изменений у детей и остеопороза у взрослых.
ФВ: табл. 0,1; 0,05; 0,005
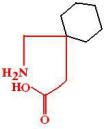
Вальпроаты. Вальпроевая кислота (Valproic acid, Dipromal, Apilepsin), натрия вальпро-
ат (Valproat sodium, Depakine). Вальпроевая кислота длительное время применялась как растворитель в составе инъекционных форм противоэпилептических препаратов. Как самостоятельный препараты начали применяться во Франции с 1969 г.
МД: Противосудорожное действие жирных кислот известно давно, оптимальным оно является у кислот с пятичленной алифатической цепью. Разветвленность цепи никоим образом не влияет на на противосудорожную активность, но значительно повышает липофильность кислоты, а
занчит и ее способность проникать через ГЭБ. В настоящее время предполагается несколько механизмов противосудорожного действия вальпроатов:
Вальпроаты блокируют фермент ГАМК-Т, которая разрушает ГАМК до янтарного полуальдегида. Кроме того, вальпроаты нарушают обратный нейрональный захват ГАМК и, в
итоге, уровень ГАМК в синапсах ЦНС резко повышается. Активируя ГАМКА-рецепторы, ГАМК открывает хлоридные каналы и ток ионов хлора в клетку вызывает гипер поляризацию мембраны и снижение возбудимости нейронов.
В относительно низких дозах ГАМК активирует К+-каналы и обеспечивает выход ионов К+ из клетки с развитием гиперполяризации нейронов и снижением их возбудимости.
Показано, что в высоких дозах вальпроаты, подобно фенитоину, задерживают натриевые каналы в инактивированном состоянии, снижая возбудимость нейронов.
ФК: После перорального приема вальпроаты хорошо всасываются (биодоступность около 80%), но пища может замедлить скорость абсорбции вальпроатов. В крови вальпроевая кислота на 90% связана с белком (главным образом альбумином), поэтому при одновременном приеме других лекарств, которые хорошо связываются с белками возможна конкуренция этих средств друг с другом и вытеснение вальпроатов из связи с белком, приводящее к возрастанию доли свободной (активной фракции) вальпроевой кислоты в крови.
Выведение вальпроатов достаточно медленное, главным образом, вальпроаты подвергаются - и -окислению в печени с последующей конъюгацией с глюкуроновой кислотой и выведением с мочой.
ФЭ: Противоэпилептический эффект – в настоящее время вальпроаты используют как средства первого ряда при различных формах эпилепсии. Особенно высока их эффективность при абсансах и миоклонус эпилепсии.
РД: Вальпроаты применяют внутрь в дозе 25-30 мг/кг/сут, при необходимости дозу повышают до 60-100 мг/кг/сут. В отличие от других противоэпилептических средств вальпроаты не оказывают седативного эффекта.
НЭ: токсичность вальпроатов достаточно низкая и в целом они хорошо переносятся пациентами.
1.Наиболее частые нежелательные эффекты – тошнота, рвота, изжога и боли в желудке.
2.При применении высоких доз возможно развитие тремора.
3.Иногда возникают обратимое увеличение массы тела, повышение аппетита, выпадение волос (алопеция).
4.Возможно развитие идиосинкразии на вальпроаты, которое проявляется тяжелым поражением печени (обычно возникает у молодых пациентов в первые 4 месяца терапии) или развитием тромбоцитопении и агранулоцитоза.
159
5.Прием вальпроатов во время беременности приводит к развитию у ребенка пороков сердца, орофарингеальных аномалий, аномалий пальцев и spina bifida.
ФВ: Вальпроевая кислота табл. 0,1 и 0,2; драже 0,15 и 0,3; капли для приема внутрь 300мг/мл флаконы по 60 мл. Вальпроат натрия таблетки по 0,3; порошок во флак. для инъекций по 0,4. Таблетки ретард, содержащие вальпроевую кислоту и ее натриевую соль по 0,3 и 0,5 (Depakine chrono). Ввиду высокой гигроскопичности таблетки должны быть защищены от действия влаги.
Габапентин (Gabapentin, Neurontin). Представляет собой молекулу ГАМК, ковалентно связанную с циклогесаном. Механизм действия: До конца не выяснен, полагают, что габапентин усиливает высвобождение ГАМК из пресинаптических окончаний и обладает слабым стимулирующим влиянием на ГАМКА-рецепторы. Под влиянием габапентина и избытка ГАМК происходит активация рецепторов и открытие хлоридных каналов. Ток ионов хлора в клетку приводит к гиперполяриза-
ции мембран и снижению активности нейронов.
ФК: Габапентин хорошо всасывается после перорального применения, выводится почками в неизмененном виде, поэтому у лиц с патологией почек должен применяться с осторожностью (т.к. его выведение понижается и концентрация габапентина в крови возрастает).
ФЭ: Габапентин оказывает противосудорожный эффект при парциальных судорогах. РД: Терапевтические дозы составляют 1800-3600 мг в день.
НЭ: Сонливость, головокружение, атаксия, головная боль, тремор.
ФВ: капс. 100, 300 и 400 мг
Бензодиазепины. Клоназепам (Clonazepam, Antelepsin). Диазепам (Diazepam, Relanium, Seduxen, Valium). Бензодиазепины – достаточно большая и интенсивно развивающаяся группа лекарственных средств. В терапии эпилепсии определенную роль играют только 6 представителей этой группы – диазепам, клоназепам, лоразепам, нитразепам, клоразепат и клобазам. Ниже мы рассмотрим только диазепам и клоназепам, которые применяются как противосудорожные средства в отечественной медицине.
МД: Бензодиазепины обладают двойным механизмом противосудорожного действия:
Бензодиазепины связываются с бензодиазепиновыми участками аллостерического центра
ГАМКА-рецептор хлоридного ионофорного комплекса и увеличивают сродство ГАМКА рецептора к эндогенному медиатору ГАМК. В итоге, даже ничтожно малые количества
ГАМК способны активировать ГАМКА-рецептор и вызвать открытие хлоридного канала. При этом в отличие от барбитуратов, бензодиазепины увеличивают частоту открытия канала на ГАМК, а не его длительность. Ток ионов хлора через каналы внутрь нейронов коры головного мозга приводит к гиперполяризации мембраны и угнетению активности эпилептогенного очага. Ток ионов хлора через каналы внутрь интернейронов спинного мозга приводит к гиперполяризации их мембраны и угнетению полисинаптических мышечных рефлексов, что вызывает дополнительную миорелаксацию.
В больших дозах бензодиазепины способны пролонгировать инактивированное состояние натриевых каналов.
ФК: И диазепам, и клоназепам достаточно хорошо и полно абсорбируются из ЖКТ.
При лечении эпилепсии используют пероральное применение клоназепама (т.к. он обладает длительным действием) и внутривенное введение диазепама (т.к. он обладает непродолжительным действием). Внутримышечное введение бензодиазепинов применять не рекомендуется, т.к. в мышечной ткани они образуют плохорастворимое депо, из которого всасываются плохо и с непостоянной скоростью. Метаболизм диазепама и клоназепама происходит в печени, при этом происходит восстановление нитрогруппы до аминогруппы с потерей фармакологической активности этих средств.
ФЭ:
1.Противоэпилептический эффект. Каждый из бензодиазепинов имеет свой профиль противосудорожной активности. Диазепам эффективен при лечении grand mal и купировании
160
эпилептического статуса, тогда как клоназепам наиболее эффективен при абсансах и миоклонус эпилепсии.
2.Седативный (успокаивающий) эффект – проявляется подавлением реакции на постоянные раздражители, снижением уровня спонтанной активности и мышления.
3.Транквилизирующий эффект (атарактический эффект) – устранение чувства тревоги, страха, беспокойства.
4.Снотворный эффект – в отличие от фенобарбитала бензодиазепины минимально изменяют структуру сна, они облегчают засыпание и удлиняют время сна.
5.Миорелаксирующий эффект – бенхзодиазепины снижают мышечный тонус за счет подавления полисинаптических рефлексов спинного мозга.
РД: Прием клоназепама начинают с минимальных доз (ввиду сильного седативного
эффекта), постепенно повышая их до 0,1-0,2 мг/кг/сут в 1-2 приема. Диазепам назначают по 0,2-0,5 мг/кг внутривенно для купирования эпилептического статуса, инъекции повторяют по мере необходимости (максимально допустимая доза 100 мг/сут). У детей иногда применяют ректальное введение диазепама.
НЭ:
1.Сонливость, летаргия, антероградная амнезия (нарушения памяти на текущие события), головокружение. Эти симптомы особенно выражены вначале терапии бензодиазепинами.
2.Мышечная слабость, атаксия (нарушение походки и поддержания положения тела). Данные эффекты чаще возникают у пожилых лиц.
3.К противосудорожному эффекту бензодиазепинов с течением времени развивается толерантность.
4.Развитие лекарственной зависимости по бензодиазепиновому типу, с абстинентным синдромом при прекращении приема (нарушения поведения, немотивированная агрессивность, неспособность к концентрации внимания, необъяснимый страх, тревога, анорексия или повышения аппетита, повышение тонуса мышц, бессонница).
5.У пожилых лиц и подлных пациентов возможно возникновение синдрома «ночных апноэ» (остановок дыхания на 20-30 сек с всхрапыванием).
ФВ: Клоназепам табл. 0,25 и 1 мг. Диазепам ампулы 0,5% раствора по 2 мл, табл. по 2, 5,
10 мг, капс. по 15 мг
Ламотриджин (Lamotrigine, Lamictal). Производное фенилтриазина. Был создан случайно, на основе ложного представления о том, что противосудорожная активность фени-
тоина обусловлена его антагонизмом к фолиевой кислоте.
МД: Показано, что ламотриджин тормозит выделение из пресинаптических окончаний возбуждающих аминокислот – аспарагиновой и глютаминовой. В итоге, не активируются NMDA и AMPA рецепторы постсинаптической мембраны, сопряженные с Ca2+ и Na+-каналами соответственно. Нарушение тока ионов натрия и кальция в клетку приводит к нарушению генерации кратковременных и длительных возбуждающих потенциалов действия, снижению активности нейрона.
Недавно было показано, что подобно фенитоину ламотриджин способен пролонгировать инактивированное состояние натриевых каналов.
ФК: Ламотриджин хорошо всасывается после перорального введения, период его полуэлиминации около 24 часов, что позволяет применять средство 1 раз в день.
ФЭ: Ламотриджин оказывает противосудорожное действие при парциальных припадках, а также абсансах и миоклонус эпилепсии.
РД: Применяют внутрь по 100-300 мг/сут максимально до 700 мг/сут. НЭ: головокружение, головная боль, диплопия, тошнота, сыпь.
ФЭ: табл. по 25, 50 и 100 мг, табл. жевательные по 5, 25 и 100 мг