

14. Переваривание и всасывание нуклеопротеинов.
Нуклеопротеины – сложные белки, состоящие из белков и нуклеиновых кислот. Существует два типа нуклеопротеинов, которые отличаются друг от друга по составу, размерам и физико-химическим свойствам: дезоксирибонуклеопротеины (ДНП) и рибонуклеопротеины (РНП).
Нуклеопротеины пищи подвергаются перевариванию в ЖКТ, образуя ряд низкомолекулярных продуктов, всасывающихся в тонком кишечнике.
1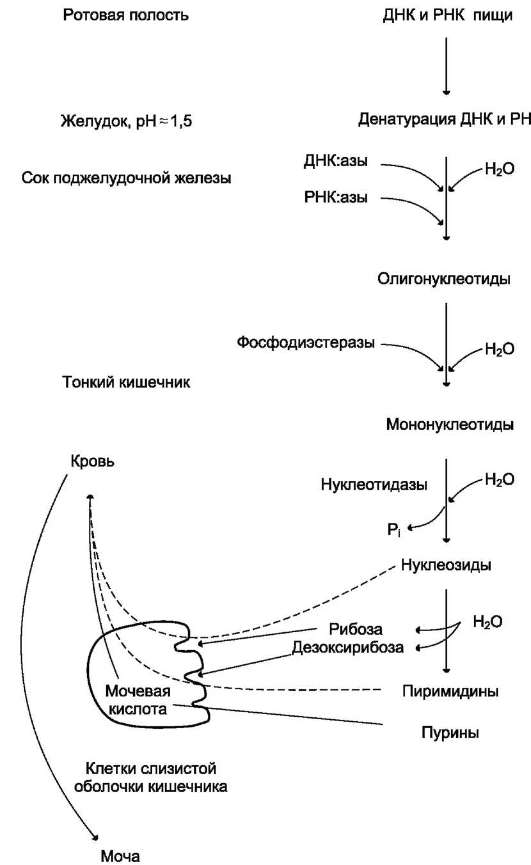 этап
– это отщепление нуклеиновой кислоты
от белковой части нуклеопротеина. Этот
разрыв связи между белком и простетической
группой происходит как в желудке , так
и в кишечнике. В желудке этот процесс
происходит под действием пепсина и HCl
желудочного сока, в кишечнике – под
действием трипсина. Далее полинуклеотидная
часть этих молекул гидролизуется в
кишечнике до мононуклеотидов.
этап
– это отщепление нуклеиновой кислоты
от белковой части нуклеопротеина. Этот
разрыв связи между белком и простетической
группой происходит как в желудке , так
и в кишечнике. В желудке этот процесс
происходит под действием пепсина и HCl
желудочного сока, в кишечнике – под
действием трипсина. Далее полинуклеотидная
часть этих молекул гидролизуется в
кишечнике до мононуклеотидов.
2 этап - расщепление нуклеиновых кислот, содержащихся в пище, происходит в результате переваривания в тонком кишечнике под действием ДНК-азы и РНК-азы панкреатического сока, которые, будучи эндонуклеазами, гидролизуют макромолекулы до олигонуклеотидов. Последние под действием фосфодиэстераз панкреатической железы расщепляются до смеси 3'- и 5'-мононуклеотидов. Нуклеотидазы и неспецифические фосфатазы гидролитически отщепляют фосфатный остаток нуклеотидов и превращают их в нуклеозиды, которые либо всасываются клетками тонкого кишечника, либо расщепляются нуклеозидфосфорилазами кишечника с образованием рибозо- или дезоксирибозо-1-фосфата, пуриновых и пиримидиновых оснований.
Всасывание продуктов гидролиза нуклеиновых кислот происходит в виде нуклеотидов и нуклеозидов, а также в виде азотистых оснований, пентозы и остатка фосфорной кислоты.
Пищевые пурины и пиримидины не являются незаменимыми пищевыми факторами и очень мало используются для синтеза нуклеиновых кислот тканей. В энтероцитах обнаружена высокая активность ксантиноксидазы - фермента, который большую часть пуринов, поступающих в клетки, превращает в мочевую кислоту, удаляющуюся с мочой. Пиримид. основания, не успевшие поступить в энтероциты, под действием микрофлоры кишечника расщепляются до NH3, CO2, β-аланина и β-аминоизобутирата.
15. Распад пуриновых нуклеотидов.
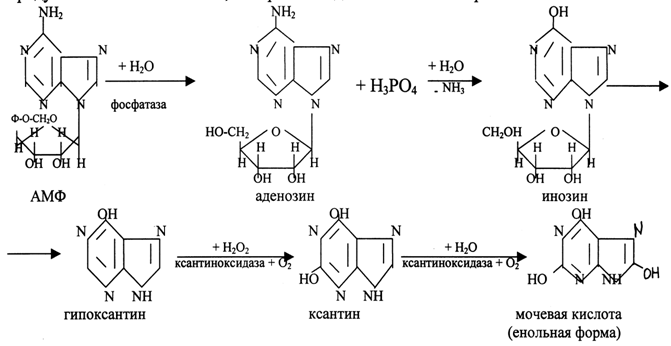
Распад пуриновых нуклеотидов.
|
Аденозин и гуанозин, которые образуются при гидролизе пуриновых нуклеотидов, подвергаются ферментативному распаду с образованием конечного продукта – мочевой кислоты, которая выводится с мочой из организма.
|
Распад пиримидиновых нуклеотидов.
|
Начальные этапы этого процесса катализируются специф. ферментами. Конечные продукты: СО2, NН3, мочевина, β-аланин, β-аминоизомасляная кислота. β-аланин используется для синтеза дипептидов мышц – карнозина и ансерина или выделяется с мочой.
|

16. Заболевания, связанные с нарушением обмена нуклеотидов: подагра, синдром Леша – Нихена.
Гиперурикемия – повышение в плазме крови концентрации мочевой кислоты. Вследствие гиперурикемии может развиться подагра.
Подагра – заболевание, вызванное нарушением обмена нуклеиновых кислот. В хрящах, сухожилиях, в суставных сумках, иногда в почках, коже, мышцах откладываются кристаллы мочевой кислоты и уратов. Вокруг этих отложений образуется воспаление и грануляционный вал, который окружает омертвевшую ткань, при этом образуются подагрические узлы - тофусы (в суставах пальцев рук, ног, в хрящах ушной раковины), что сопровождается деформацией и болезненностью пораженных суставов. К характерным признакам подагры относятся повторяющиеся приступы острого воспаления суставов (чаще всего мелких) – острого подагрического артрита. Обычно больные склонны к атеросклерозу и гипертонии. В их крови наблюдается большая концентрация мочевой кислоты – гиперурикемия. В течение нескольких дней перед приступом подагры увеличивается выделение воды и хлорида натрия с мочой, т.е. сдвигается водно-солевой баланс. Вследствие этого возрастает концентрация мочевой кислоты в крови и отложение ее в тканях.
Как правило, подагра генетически детерминирована и носит семейный характер. Она вызвана нарушениями в работе фосфорибозилдифосфата (ФРДФ) синтетазы или гипоксантингуанин- или аденинфосфорибозилтрансфераз.
К другим характерным проявлениям относят нефропатию, при которой наблюдают образование уратных камней в мочевыводящих путях.
Синдром Леша-Нихена – тяжелая форма гиперурикемии, которая наследуется как рецессивный признак, сцепленный с Х-хромосомой. Проявляется только у мальчиков. Кроме симптомов подагры наблюдаются церебральные параличи, нарушение интеллекта, попытки наносить себе раны (укусы губ, пальцев). Связана болезнь с дефектом фермента гипоксантин-гуанин-фосфорибозилтрансферазы, которая катализирует превращение гипоксантина и гуанина в гуанинимонофосфат (ГМФ), поэтому они превращаются в мочевую кислоту. В первые месяцы жизни неврологические расстройства не обнаруживаются, но на пеленках отмечают розовые пятна, вызванные присутствием в моче кристаллов мочевой кислоты. При отсутствии лечения больные погибают в возрасте до 10 лет из-за нарушения функции почек.
Основной препарат для лечения гиперурикемии – аллопуринол (структурный аналог гипоксантина).