

Modern Organocopper Chemistry
.pdf1785 Copper(I)-mediated 1,2- and 1,4-Reductions
glovebox. In the glovebox, NaOt-Bu (5 mg, 0.05 mmol) and CuCl (5 mg, 0.05 mmol) were weighed into a vial. The toluene solution of the chiral bisphosphine was added by pipette to the vial to dissolve solids and the resulting solution was then transferred back into the Schlenk tube. The Schlenk tube was removed from the glovebox, the solution was stirred for 10–20 min, and PMHS (0.063 mL, 1.05 mmol) was added to the solution with argon purging. The resulting solution turned reddish orange in color. The solution was then cooled to the specified temperature. The a; b-unsaturated ketone (1.0 mmol) was added to the reaction mixture with argon purging and the resulting solution was stirred at room temperature (18–27 h). Consumption of the a; b-unsaturated ketone was monitored by GC. When the reaction was complete, the Schlenk tube was opened and water (1 mL) was added. The resulting solution was diluted with diethyl ether, washed once with water and once with brine, and back-extracted with diethyl ether. TBAF (1 mmol, 1 M in THF) was added to the combined organic extracts and the resulting solution was stirred for 3 h. The solution was then washed once with water and once with brine, back-extracted with diethyl ether, and the organic layer was dried over anhydrous MgSO4. The solvent was then removed in vacuo and the product was purified by silica column chromatography. In order to determine the ee, the product was converted into the corresponding (R,R)-2,3-dimethylethylene ketal and then analyzed by GC analysis (Chiraldex G-TA) for the diastereomeric ketals.
Intermediate silyl enol ethers can be trapped and isolated from initial conjugate reductions of enones with Stryker’s reagent, or they may be used directly in Mukaiyama-type aldol constructions (i.e, in 3-component constructions; 3-CC) [35]. Thus, in a one-pot sequence using toluene as the initial solvent and 1–5 mol% (Ph3P)CuH relative to enone, any of a number of silyl hydrides (such as PhMe2SiH, Ph2MeSiH, tetramethyldisiloxane (TMDS), or PMHS) can be employed to produce the corresponding silyl enol ether. Dilution with CH2Cl2 without isolation, followed by cooling to 78 C and introduction of an aldehyde, followed by a Lewis acid (TiCl4 or BF3 OEt2) results in good yields of aldol adducts (Eq. 5.18). Unfortunately, there is no acyclic stereocontrol (syn versus anti selectivity) in these 3-CC reactions [34b].
ð5:18Þ
Representative procedure for conjugate reduction-aldol 3-CC: 2-{Hydroxy-[1-(toluene-4- sulfonyl)-1H-indol-3-yl]-methyl}-4,4-dimethylcyclohexanone [35]
Dimethylphenylsilane (0.23 mL, 1.5 mmol, 1.5 equiv.) was added dropwise to a homogeneous, red solution of [CuH(PPh3)]6 (16.0 mg, 0.008 mmol, 5 mol% Cu) in toluene (2.0 mL) and the solution was stirred at room temperature for ca. 5 min. 4,4-Dimethylcylohexenone (0.13 mL, 1.0 mmol) was

5.4 1,2-Reductions Catalyzed by Copper Hydride 179
added dropwise to the resulting red solution, which was stirred at room temperature. After ca. 7 min, the solution had darkened to a heterogeneous brown/black. Monitoring of the reaction by TLC showed that the enone had been consumed after 3 h, forming the corresponding silyl enol ether. The solution was diluted with CH2Cl2 (5.0 mL) and added by cannula to a solution of N-tosyl-indole-3-carboxaldehyde (0.45 g, 1.5 mmol, 1.5 equiv.) and TiCl4 (1.5 mL of 1.0 M solution in CH2Cl2, 1 equiv.), in CH2Cl2 (7.0 mL) at78 C. Stirring was continued for 1 h and the reaction was quenched with saturated NaHCO3 solution (6.0 mL) at 78 C, and allowed to warm to room temperature. A blue precipitate was filtered using a Buchner funnel, and the aqueous layer was extracted with diethyl ether (3 25 mL). The combined organic portions were washed with brine (2 50 mL) and dried over anhydrous Na2SO4, and the solvent was removed in vacuo. Purification by flash chromatography (1:9 EtOAc/PE to 1:4 EtOAc/PE) a orded diastereomers as a yellow oil (combined yield 0.35 g, 82%).
5.4
1,2-Reductions Catalyzed by Copper Hydride
Reductions of non-conjugated aldehydes and ketones based on copper chemistry are relatively rare. Hydrogenations and hydrosilylations of carbonyl groups are usually e ected by transition metals such as Ti [36], Rh [37], and Ru [38], and in one case, Cu [39]. An early report using catalytic [(tol)3P]CuH in reactions with formaldehyde, in which disproportionation characteristic of a Tishchenko reaction took place, is indicative of a copper(I) alkoxide intermediate [16]. Almost two decades later, variations in the nature of the triphenylphosphine analogue (Strykers’ reagent), principally induced by introduction of alternative phosphine ligands, have resulted in remarkable changes in the chemoselectivity of this family of reducing agents [40, 41]. Although not as yet fully understood, subtle di erences even between alkyl substituents on phosphorus can bring about dramatic shifts in reactivity patterns. Changes in the composition of [(Ph3P)CuH]6 caused by ligands such as tripod (1,1,1-tris-(diphenylphosphinomethyl)-ethane), which forms a dinuclear bidentate complex (Fig. 5.3) [42], have been used by Stryker to great advantage to reduce ketones in a 1,2-fashion.
Both conjugated and non-conjugated ketones, as well as conjugated aldehydes, undergo clean 1,2-addition in the presence of CuH modified by Me2PhP (Eq. 5.19). Ketones react under an atmosphere of hydrogen over a roughly 24 hour period. The presence of t-BuOH (10–20 equiv./copper) is important for increasing catalyst life-
Fig. 5.3. Ligands tested for 1,2-reductions.

180 5 Copper(I)-mediated 1,2- and 1,4-Reductions
time, as in the corresponding cases of 1,4-reductions (vide supra), presumably by conversion of the initially formed copper alkoxide to the alcohol product in exchange for a thermally more stable [Cu(O-t-Bu)]4. This complex is then hydrogenolyzed to reform the copper hydride catalyst. In most cases, isolated olefins are untouched, as is true for dienes, esters, epoxides, alkynes, and acetals. Rates are slower in substrates bearing free alkenes, probably a consequence of d–p interactions with the metal. Acyclic conjugated enones a ord a high degree of control for generation of allylic alcohol products, with only small percentages of overreduced material formed when using PhMe2P-modified reagent. The corresponding PhEt2P-altered Stryker’s reagent, however, does not function as a catalyst for this chemistry (this is also the case with the novel biaryl P,O-ligand 12, the dimethylphosphino analog of MOP) [43], while the mixed dialkylphenyl case Me(Et)PPh is unexpectedly e ective (e.g., for b-ionone, 13: >50:1; 95% yield; Eq. 5.20).
(5.19)
(5.20)
With these new levels of appreciation of the nuances associated with CuH-phos- phine interactions, considerable fine-tuning of Stryker’s reagent is now possible. One case in point involves enone 14, which can be converted predominately into any one of three possible products (Scheme 5.6) [40].
Scheme 5.6. Selective reductions as a function of phosphine.

5.4 1,2-Reductions Catalyzed by Copper Hydride 181
General procedure for reduction of saturated ketones using [(Ph3P)CuH]6 and Me2PPh
[40]
In a glovebox, [(Ph3P)CuH]6 (1–10 mol% Cu), Me2PPh (6 equiv./Cu), and t-butanol (10–20 equiv./Cu) were combined in a Schlenk flask and dissolved in benzene. A solution of the substrate (10–100 equiv./Cu) in benzene (0.4–0.8 M in substrate) was added to this solution. The flask was sealed, removed from the drybox and, after one freeze-pump-thaw degassing cycle, placed under a slight positive pressure of hydrogen. The resulting yellow-orange homogeneous solution was allowed to stir until completion, as monitored by TLC. The reaction mixture was exposed to air, diluted with ether, and treated with a small amount of silica gel. This mixture was stirred in air for b0.5 h, filtered, concentrated in vacuo, and purified by flash chromatography. If the polarity of the product was similar to that of the residual phosphine, the crude mixture was treated with sodium hypochlorite (5% aqueous solution) and filtered through silica gel/MgSO4 prior to chromatography.
General procedure for reduction of saturated ketones using (PhMe2P)CuH produced in situ [40]
Under an inert atmosphere, a solution of the substrate in benzene was added to a slurry of freshly purified CuCl (5 mol%), Me2PPh (6 equiv./Cu), and t-butanol (10 equiv./Cu) in benzene (final concentration: 0.4–0.8 M in substrate). After degassing with one freeze-pump-thaw cycle, the suspension was placed under a slight positive pressure of hydrogen and allowed to stir until completion, as monitored by TLC. The product was isolated and purified as described above.
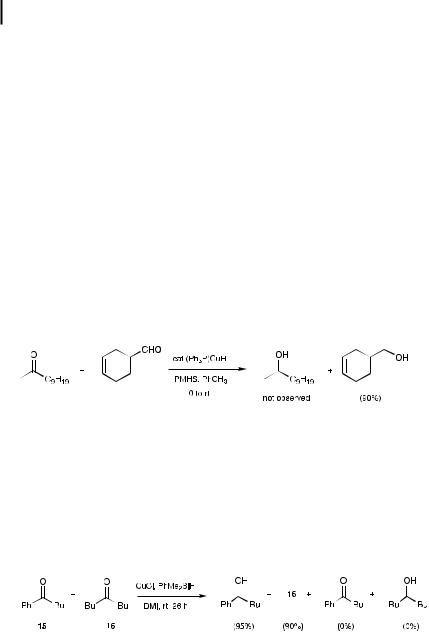
Further alterations in the above reaction conditions, notably the replacement of H2 with various silanes as the hydride source, results in a net hydrosilylation of nonconjugated aldehydes and ketones [44]. The catalytic (PPh3)CuH/excess R3SiH combination is highly e ective at converting aldehydes directly into protected primary alcohols, with silanes ranging from PhMe2SiH – which produces a relatively labile silyl ether – to Hanessian’s especially hydrolytically stable t-BuPh2Si derivatives [45], all from the corresponding precursor silanes (Eq. 5.21). Levels of CuH used tend to be in the 1–3 mol% range, although from the few cases studied to date, one tenth as much may be su cient to drive the reaction to completion. The more reactive PMHS [30] appears to be the ideal choice of silane for catalyst usage in the <1 mol% category, although the use of this polymeric hydride source necessitates workup under basic conditions.
ð5:21Þ
1825 Copper(I)-mediated 1,2- and 1,4-Reductions
Representative 1,2-reduction/silylation of an aldehyde, giving (2-bromobenzyloxy)- diphenylmethylsilane [44]
A dried 25 mL flask with a rubber septum top was flushed with argon and charged with [PPh3(CuH)]6 (53 mg, 0.162 mmol), as a red solid. Toluene
(5.4 mL) was added, followed by neat diphenylmethylsilane (1.4 mL, 7.0 mmol), resulting in a homogeneous red solution. In a second dry, argonflushed vessel (10 mL), fitted with a rubber septum, 2-bromobenzaldehyde (0.63 mL, 5.4 mmol) and toluene (4 mL) were mixed together, and the solution was transferred by cannula, with stirring, into the solution (at room temperature) of copper reagent and silane. The reaction mixture was monitored by TLC (elution with 5% diethyl ether/hexane, Rf ¼ 0:74); the aldehyde was consumed after 30 min. The reaction was filtered through a pad of Celite/charcoal, washed with EtOAc (2 15 mL), and the filtrate concentrated to an oil in vacuo. Kugelrohr distillation (168 C, 0.2–0.3 Torr) yielded the title compound as a colorless oil (1.98 g, 95%).
Ketones take considerably longer to reduce than aldehydes (10–24 h), although yields are not compromised. Di erences in reactivity toward aldehydes and ketones can be used to advantage, with highly chemoselective reduction occurring at the aldehyde in the presence even of a methyl ketone (Eq. 5.22) [44].
ð5:22Þ
In situ production of phosphine-free CuH from CuCl or CuOAc (0.3–1.0 equivalents), in the presence of an excess of PhMe2SiH in DMI at room temperature, displays a remarkable preference for reductions of aryl ketones (e.g., 15) over aliphatic ones such as 16 (Eq. 5.23) [46]. Reactions require a day or more to reach completion, concentrations of 0.5 M notwithstanding, but yields have been uniformly good (77–88%) for the few cases examined. Aldehydes, however, show no such selectivity and are reduced to the corresponding primary alcohols, albeit in high yields.
ð5:23Þ
5.5
Heterogeneous CuH-Catalyzed Reductions
Catalysts such as copper chromite, first prepared and utilized for carbonyl 1,2- reductions back in 1931 [47], have given way to more modern reagents for e ecting

5.5 Heterogeneous CuH-Catalyzed Reductions 183
related transformations under heterogeneous conditions. Ravasio first described Cu/Al2O3 in steroid reductions (steroid-4-en-3-ones), examining the regioselectivities, stereoselectivities, and chemoselectivities of this supported reductant at 60 C under a hydrogen pressure of one atmosphere [48]. A follow-up study by that group, described in 1996, promotes the more generally useful Cu/SiO2 [48]. Under an atmosphere of H2 at 90 C in toluene, this catalyst e ects 1,4-reductions of conjugated enones while leaving isolated olefins intact. Although the preparation of the catalyst is fairly involved (cf. the procedure below), the method results in excellent levels of conversion, and high yields of the corresponding ketones. The featured example in this work is that of b-ionone, from which the desired keto product, reflecting reduction of the a; b-site, was provided with high levels of regiocontrol (Eq. 5.24). Removal of the catalyst by filtration, followed by reactivation at 270 C, essentially did not result in any change in selectivity after four consecutive cycles. These reactions are believed to involve CuH, generated on the surface of pyrogenic silica.
ð5:24Þ
Catalyst preparation [49]
Concentrated NH4OH was added to a solution of Cu(NO3)2 3H2O (25 mL, 160 g/L) until pH ¼ 9 was reached, the support (silica, 10 g) was then added, and the mixture was slowly diluted to 3 L in order to allow hydrolysis of the Cu[NH3]þþ4 complex and deposition of the finely dispersed product to occur. The solid was separated by filtration, washed with water, dried overnight at 120 C, and calcined in air at 350 C for 3 hours. In this way, 8% Cu samples, 308 m2/g BET surface area, were obtained. The catalyst was reduced with H2 at 270 C at atmospheric pressure, the water formed being removed under reduced pressure, before the hydrogenation reaction.
Experimental conditions
The substrates (2 mmol) were dissolved in toluene (12 mL) and the solution was transferred under H2 into a glass reaction vessel in which the catalyst (0.3 g) had been reduced previously. Reactions were carried out at 90 C and at atmospheric pressure, with magnetic stirring, the final charge of hydrogen being adjusted to 1 atm with a mercury leveling bulb, and monitored by withdrawing 20 mL samples through a viton septum and analyzing them by capillary GLC. After completion, the catalyst was filtered o , the solvent removed, and the reaction mixture analyzed by NMR. Superatmospheric pressure (1.5–5 atm) could conveniently be used to speed up the reaction without loss in selectivity when higher substrate/Cu ratios were used. For the recycling tests, the catalyst was washed with diethyl ether, dried, and reactivated at 270 C.

184 5 Copper(I)-mediated 1,2- and 1,4-Reductions
A fascinating study on the surface science of copper hydride on SiO2, as well as on Al2O3, ceria (cerium oxide), and ZnO, has appeared [50]. Pure, yet thermally unstable, CuH can be precipitated as a red-brown solid from aqueous cupric sulfate and hypophosphorous acid in the presence of H2SO4, and has been characterized by powder X-ray di raction (PXRD) (Eq. 5.25). Transmission electron microscopy (TEM) data suggest that it is most stable when deposited on acidic SiO2.
(5.25)
5.6
Overview and Future Developments
Although many variations on reagents bearing hydride ligated to copper(I) have been developed, it was the advent of Stryker’s reagent that provided a well defined, easily handled, and crystalline source of CuH. This hexameric copper hydride, [(Ph3P)CuH]6, has been enthusiastically embraced by the synthetic community as a highly reliable means of e ecting fundamental conjugate reductions of unsaturated aldehydes, ketones, and esters. Unlike the procedures previously in use, in which presumed ate complexes of CuH required manipulations of multiple reagents and gave rise to highly basic species, (Ph3P)CuH is relatively non-basic and is available commercially, or can be readily prepared in multigram quantities. Moreover, when stored under an inert atmosphere, it can last for months without significant decomposition. That (Ph3P)CuH derives from readily accessible and inexpensive precursors is a bonus, and as it is regarded as a base metal catalyst, in association with either molecular hydrogen or silanes as sources of stoichiometric hydride, the economics involved in its use are highly favorable. Also not to be overlooked among the virtues of (Ph3P)CuH is its tolerance to moisture, as well as many to functional groups – including isolated, unsaturated carbon-carbon bonds – which otherwise preclude normal modes of catalytic hydrogenation. The noteworthy impact exerted by various achiral monodentate and bidentate phosphine ligands on CuH reactions can be used to tremendous advantage in controlling resulting regioselectivities and chemoselectivities. Replacement of the PPh3 in Stryker’s reagent with selected chiral, nonracemic bidentate phosphines has enabled enantioselective 1,4-reductions to be achieved. Still more recently, the 1,2-addition mode of Stryker’s reagent has been evolving rapidly. These reactions have similarly proven to be quite e ective under conditions catalytic in CuH. Further recognition and greater appreciation of such elements of reactivity and selectivity, associated with both the 1,2- and the 1,4-reduction patterns of (Ph3P)CuH, are likely to give rise to future improvements, new methodologies, and synthetic applications.
ð5:26Þ
References 185
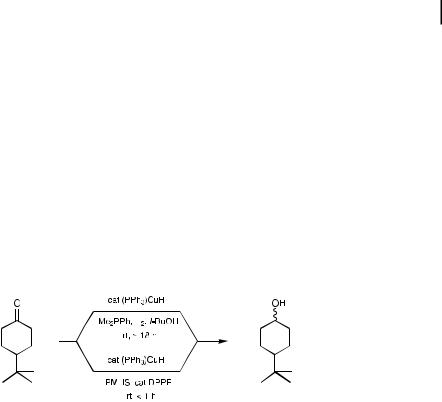
An aldimine reduction already ‘‘in the pipeline’’ has been tested using catalytic Stryker’s reagent along with various silanes, the preliminary data suggesting that such 1,2-additions do indeed take place, albeit far more slowly that those on the corresponding carbonyl derivatives (Eq. 5.26) [51]. In line with observations made concerning the e ects of phosphines on CuH [40, 41], a remarkable rate enhancement has also been noted in ketone hydrosilylations under the influence either of racemic BINAP or DPPF (bis(diphenylphosphino)ferrocene). Thus, while 4-t- butylcyclohexanone takes a day to be reduced when catalytic (PPh3)CuH is used with either H2 [40, 41] or PMHS [44], simple addition of either of these bidentate ligands results in complete conversion in less than one hour at identical concentrations (Scheme 5.7) [44]. This key observation has generated considerable enthusiasm for development of a highly e ective method for asymmetric hydrosilylations [52] of aryl ketones using catalytic CuH ligated by a nonracemic bidentate phosphine (Roches’ 3.5-xyl-MEO-BIPHEP) [53]. It thus seems reasonable to conclude that the story of reductions by CuH in organic synthesis, whether under homogeneous or heterogeneous conditions, is far from complete.
Scheme 5.7. E ect of DPPF on reductions with Stryker’s reagent.
References
1M. S. Kharasch, P. O. Tawney, J. Am. Chem. Soc. 1941, 63, 2308–2315.
2H. Gilman, R. G. Jones, L. A. Woods, J. Org. Chem. 1952, 17, 1630–
1634.
3A. Wurtz, Ann. Chim. Phys. 1844, 11, 250–251.
4 H. Muller, A. Bradley, J. Chem. Soc.
1926, 1669–1673.
5(a) G. M. Whitesides, E. R. Stredronsky, C. P. Casey, J. San
Filippo, J. Am. Chem. Soc. 1970, 92, 1426–1427. (b) G. M. Whitesides, J. San Filippo, E. R. Stredronsky,
C. P. Casey, J. Am. Chem. Soc. 1969,
91, 6542–6544. (c) G. M. Whitesides, C. P. Casey, J. Am. Chem. Soc. 1966,
88, 4541–4543.
6(a) M. R. Churchill, S. A. Bezman, J. A. Osborn, J. Wormald, Inorg. Chem. 1972, 11, 1818–1825. (b) S. A. Bezman, M. R. Churchill, J. A. Osborn, J. Wormald, J. Am. Chem. Soc. 1971, 93, 2063–2065. For an early cryoscopic study on CuH-phosphine interactions, see J. A. Dilts, D. F. Shriver, J. Am. Chem. Soc. 1969, 91, 4088–4091.
7For typical uses of these groups as ‘dummy ligands’ see (a) B. H. Lipshutz, in Organometallics in Synthesis: A Manual, Schlosser,
M. (ed.), Wiley, 1994, pp 283– 382. (b) B. H. Lipshutz, S. Sengupta, Org. React. 1992, 41, 135–631.
186 |
5 Copper(I)-mediated 1,2- and 1,4-Reductions |
|
|
|
|
|
R. K. Boeckman, R. Michalak, J. |
|
6797. (b) B. H. Lipshutz, J. Keith, |
8 |
|
|||
|
|
Am. Chem. Soc. 1974, 96, 1623–1625. |
|
D. J. Buzard, Organometallics 1999, |
9 |
T. Yoshida, E.-I. Negishi, J. Chem. |
|
18, 1571–1574. |
|
|
|
Soc. Chem. Comm. 1974, 762–763. |
22 |
H. Tanaka, Y. Yamaguchi, S.-I. |
10 |
E. C. Ashby, J.-J. Lin, A. B. Goel, |
|
Sumida, M. Kuroboshi, M. |
|
|
|
J. Org. Chem. 1978, 43, 183–188. |
|
Mochizuki, S. Torii, J. Chem. Soc., |
11 |
(a) M. F. Semmelhack, R. D. |
|
Perkin Trans. 1 1999, 3463–3468. |
|
|
|
Stauffer, J. Org. Chem. 1975, 40, |
23 |
H. Ito, T. Ishizuka, K. Arimoto, |
|
|
3619–3621. (b) M. F. Semmelhack, |
|
K. Miura, A. Hosomi, Tetrahedron |
|
|
R. D. Stauffer, A. Yamashita, J. |
|
Lett. 1997, 38, 8887–8890. |
|
|
Org. Chem. 1977, 42, 3180–3188. See |
24 |
A. Mori, A. Fujita, H. Kajiro, Y. |
|
|
also M. E. Osborn, J. F. Pegues, L. A. |
|
Nishihara, T. Hiyama, J. Chem. Soc., |
|
|
Paquette, J. Org. Chem. 1980, 45, |
|
Chem. Commun. 1997, 2159–2160. |
|
|
167–168. M. E. Osborn, S. Kuroda, |
25 |
A. Mori, A. Fujita, H. Kajiro, |
|
|
J. L. Muthard, J. D. Kramer, P. |
|
Y. Nishihara, T. Hiyama, |
|
|
Engel, L. A. Paquette, J. Org. Chem. |
|
Tetrahedron, 1999, 55, 4573–4582. |
|
|
1981, 46, 3379–3388. |
26 |
(a) T. Tsuda, H. Satomi, T. Hayashi, |
12 |
D. L. Comins, A. H. Abdulla, J. Org. |
|
T. Saegusa, J. Org. Chem. 1987, 52, |
|
|
|
Chem. 1984, 49, 3392–3394. |
|
439–443. (b) T. Tsuda, T. Hayashi, |
13 |
W. S. Mahoney, D. M. Brestensky, |
|
H. Satomi, T. Kawamoto, T. Saegusa, |
|
|
|
J. M. Stryker, J. Am. Chem. Soc. |
|
J. Org. Chem. 1986, 51, 537–540. |
|
|
1988, 110, 291–293. |
27 |
A. R. Daniewski, J. Kiegiel, J. Org. |
14 |
Listed as ‘‘hydrido |
|
Chem. 1988, 53, 5534–5535. A. R. |
|
|
|
(triphenylphosphine)copper(I) |
|
Daniewski, J. Kiegiel, J. Org. Chem. |
|
|
hexamer’’; Aldrich catalog a36497-5. |
|
1988, 53, 5535–5538. E. J. Corey, |
15 |
D. M. Brestensky, D. E. Huseland, |
|
A. X. Huang, J. Am. Chem. Soc. 1999, |
|
|
|
C. McGettigan, J. M. Stryker, |
|
121, 710–714. |
|
|
Tetrahedron Lett. 1988, 29, 3749–3752. |
28 |
A. R. Daniewski, W. Liu, J. Org. |
16 |
G. V. Goeden, K. G. Caulton, J. Am. |
|
Chem. 2001, 66, 626–628. |
|
|
|
Chem. Soc. 1981, 103, 7354–7355. |
29 |
B. H. Lipshutz, J. Keith, P. Papa, |
17 |
T. M. Koenig, J. F. Daeuble, |
|
R. Vivian, Tetrahedron Lett. 1998, 39, |
|
|
|
D. M. Brestensky, J. M. Stryker, |
|
4627–4630. |
|
|
Tetrahedron Lett. 1990, 31, 3237–3240. |
30 |
N. J. Lawrence, M. D. D. Drew, |
18 |
P. Chiu, B. Chen, K. F. Cheng, |
|
S. M. Bushell, J. Chem. Soc., Perkin |
|
|
|
Tetrahedron Lett. 1998, 39, 9229–9232. |
|
Trans. 1 1999, 3381–3391. |
19 |
(a) W. S. Mahoney, J. M. Stryker, J. |
31 |
Y. Moritani, D. H. Appella, V. |
|
|
|
Am. Chem. Soc. 1989, 111, 8818–8823. |
|
Jurkauskas, S. L. Buchwald, J. Am. |
|
|
(b) W. S. Mahoney, J. M. Stryker, |
|
Chem. Soc. 2000, 122, 6797–6798. |
|
|
In Catalysis in Organic Synthesis, |
32 |
D. H. Appella, Y. Moritani, |
|
|
Pascoe, W. E. (ed.), Marcel Dekker, |
|
R. Shintani, E. M. Ferreira, S. L. |
|
|
New York, 1992, pp. 29–44. (c) J. F. |
|
Buchwald, J. Am. Chem. Soc. 1999, |
|
|
Daueble and J. M. Stryker, |
|
121, 9473–9474. |
|
|
‘‘Highly Chemoselective Catalytic |
33 |
(a) R. Schmid, M. Cereghetti, |
|
|
Hydrogenation of Polar Unsaturation |
|
B. Heiser, P. Schonholzer, H.-J. |
|
|
Using Cu(I) Complexes and |
|
Hansen, Helv. Chim. Acta 1988, 71, |
|
|
Hydrogen,’’ in Catalysis of Organic |
|
897–929. (b) A. Knierzinger, |
|
|
Reactions, Scaros, M. and Prunier, |
|
P. Schonholzer, Helv. Chim. Acta |
|
|
M. L. (eds.), (Chem. Ind. 62); Marcel |
|
1992, 75, 1211–1220. |
|
|
Dekker, New York, 1995, 235–248. |
34 |
(a) C. Lorenz, U. Schubert, Chem. |
20 |
B. H. Lipshutz, C. S. Ung, S. |
|
Ber. 1995, 128, 1267–1269. (b) B. L. |
|
|
|
Sengupta, Synlett 1989, 63–66. |
|
Pagenkopf, J. Kru¨ger, A. |
21 |
(a) B. H. Lipshutz, K. L. Stevens, B. |
|
Stojanovic, E. M. Carreira, Angew. |
|
|
|
James, J. G. Pavlovich, J. P. Snyder, |
|
Chem. 1998, 110, 3312–3314; Angew. |
|
|
J. Am Chem. Soc. 1996, 118, 6796– |
|
Chem. Int. Ed. 1998, 37, 3124–3126. |
|
|
|
References |
187 |
|
|
|
|
|
|
(c) H. Ito, T. Ishizuka, T. Okamura, |
43 |
Y. Uozumi, A. Tanahashi, S.-Y. Lee, |
|
|
H. Yamanaka, J. Tateiwa, M. |
|
T. Hayashi, J. Org. Chem. 1993, 58, |
|
|
Sonoda, A. Hosomi, J. Organomet. |
|
1945–1948. |
|
|
Chem. 1999, 574, 102–106. |
44 |
B. H. Lipshutz, W. Chrisman, K. |
|
35 |
B. H. Lipshutz, W. Chrisman, |
|
Noson, J. Organomet. Chem. 2001, |
|
|
K. Noson, P. Papa, J. A. Sclafani, |
|
624, 367. |
|
|
R. W. Vivian, J. Keith, Tetrahedron |
45 |
S. Hanessian, P. Lavallee, Can J. |
|
|
2000, 56, 2779–2788. |
|
Chem. 1975, 53, 2975–2977. |
|
36 |
(a) J. Yun, S. L. Buchwald, J. Am. |
46 |
H. Ito, H. Yamanaka, T. Ishizuka, |
|
|
Chem. Soc. 1999, 121, 5640–5644. |
|
J. Tateiwa, A. Hosomi, Synlett 2000, |
|
|
(b) S. C. Berk, K. A. Kreutzer, |
|
479–482. |
|
|
S. L. Buchwald, J. Am. Chem. |
47 |
(a) H. Adkins, R. Connor, J. Am. |
|
|
Soc. 1991, 113, 5093–5094. |
|
Chem. Soc. 1931, 53, 1091–1095. |
|
37 |
(a) I. Ojima, T. Kogure, |
|
(b) B. Miya, F. Hoshino, I. Iwasa, |
|
|
Organometallics 1982, 1, 1390–1399. |
|
J. Catal. 1966, 5, 401–411. (c) R. |
|
|
(b) H. Nishiyama, M. Kondo, T. |
|
Hubaut, M. Daage, J. P. Bonnelle, |
|
|
Nakamura, K. Itoh, Organometallics |
|
Appl. Catal. 1986, 22, 231–241. (d) R. |
|
|
1991, 10, 500–508. (c) G. Z. Zheng, |
|
Hubaut, J. P. Bonnelle, M. Daage, |
|
|
T. H. Chan, Organometallics 1995, 14, |
|
J. Mol. Catal. 1989, 55, 170–183. |
|
|
70–79. |
48 |
(a) N. Ravasio, M. Rossi, J. Org. |
|
38 |
(a) T. Ohkuma, M. Koizumi, |
|
Chem. 1991, 56, 4329–4333. |
|
|
M. Yoshida, R. Noyori, Org. Lett. |
|
(b) N. Ravasio, M. Antenori, |
|
|
2000, 2, 1749–1751. (b) R. Noyori, |
|
M. Gargano, M. Rossi, J. Mol. Catal. |
|
|
T. Ohkuma, Pure Appl. Chem. |
|
1992, 74, 267–273. |
|
|
1999, 71, 1493–1501. (c) T. Ohkuma, |
49 |
N. Ravasio, M. Antenori, M. |
|
|
R. Noyori, Compr. Asymmetric |
|
Gargano, P. Mastrorilli, |
|
|
Catal. I–III 1999, 1, 199–246. |
|
Tetrahedron Lett. 1996, 37, 3529–3532. |
|
39 |
H. Brunner, W. Miehling, J. |
50 |
N. P. Fitzsimons, W. Jones, P. J. |
|
|
Organomet. Chem. 1984, 275, C17– |
|
Herley, Catal. Lett. 1992, 15, 83–94. |
|
|
C21. |
51 |
B. H. Lipshutz, A. B. Reed, K. |
|
40 |
J.-X. Chen, J. F. Daeuble, D. M. |
|
Noson, unpublished work. |
|
|
Brestensky, J. M. Stryker, |
52 |
H. Nishiyama, K. Itoh, In Catalytic |
|
|
Tetrahedron 2000, 56, 2153–2166. |
|
Asymmetric Synthesis; Ojima, I. (ed.), |
|
41 |
J.-X.Chen,J.F. Daeuble,J.M.Stryker, |
|
VCH, New York, 2000, Chapt. 2 and |
|
|
Tetrahedron 2000, 56, 2789–2798. |
|
references therein. |
|
42 |
G. V. Goeden, J. C. Huffman, |
53 |
B. H. Lipshutz, K. Noson, W. |
|
|
K. G. Caulton, Inorg. Chem. 1986, |
|
Chrisman, J. Am. Chem. Soc., in |
|
|
25, 2484–2485. |
|
press. |
|