

Modern Organocopper Chemistry
.pdf148 4 Copper-mediated Addition and Substitution Reactions of Extended Multiple Bond Systems
example, the chiral cuprate 8, containing a Scho¨llkopf bislactim ether moiety, was used in the first total synthesis of the antimycotic dipeptide chlorotetaine (10; Eq. 4.4) [12d]. Although the nucleophilic addition to dienone 7 in this case did not proceed regioselectively, furnishing only a 63:37 mixture of the 1,6- and 1,4-adducts, the former compound was successfully converted over several steps into diastereomerically and enantiomerically pure chlorotetaine (10).
ð4:4Þ
While copper-catalyzed Michael additions to acceptor-substituted dienes using Grignard reagents as nucleophiles were reported even earlier than the corresponding additions of (stoichiometric) organocuprates, the former transformations have largely been restricted to the synthesis of steroid hormones. In this context, in addition to tetrahydro-3H-naphthalen-2-ones, which were used as model substrates for doubly unsaturated steroids [13, 14], estradiol derivatives bearing an alkyl chain in the 7a-position are especially interesting target molecules, due to their high affinity for and specificity towards estrogen receptors [15, 16]. These unsaturated steroids may thus be particularly useful for the treatment of mammary tumors (breast cancer) [15]. As regards preparative aspects, however, the nucleophilic 1,6- addition to doubly unsaturated D4;6-steroids should proceed not only with the desired regioselectivity [13, 14, 15b, 16], but also in a diastereoselective manner, since only the 7a isomers are e ective enzyme inhibitors [15b]. Although the diastereoselectivity of the copper-catalyzed 1,6-addition of methyl Grignard reagents to D4;6-steroids may be dependent on the substitution pattern of the substrate [13a], general preference for attack from the a side has frequently been observed [13]. Wieland and Auner [13e], for example, reported an a selectivity of 90% in the cop- per-catalyzed 1,6-addition of MeMgBr to dienone 11 (Eq. 4.5). The product 12 was converted over several steps into 7a-methylestrone (13), a precursor of several highly active steroidal hormones.

4.2 Copper-mediated Addition Reactions to Extended Michael Acceptors 149
ð4:5Þ
In contrast to this, the introduction of longer alkyl chains with the aid of copperpromoted 1,6-addition reactions to D4;6-steroids normally proceeds with unsatisfactory a:b ratios [15b, 16]. In some cases, improvement of the diastereoselectivity by ‘‘fine tuning’’ of the reaction conditions has been possible. The ratio of the epimeric products 15 and 16 in the copper-catalyzed 1,6-addition of 4-pentenylmagnesium bromide to dienone 14, for example, was improved from 58:42 to 82:18 by adjust-
. 4.6) [16f ].
ð4:6Þ

150 4 Copper-mediated Addition and Substitution Reactions of Extended Multiple Bond Systems
Aberrant behavior, however, has been observed when using bicyclic tetrahydro-3H- naphthalen-2-ones as Michael acceptors: the 1,6-addition of cyano-Gilman cuprates or Grignard reagents (catalyzed by copper arene thiolate 18) proceeds with high trans selectivity, irrespective of the transferred group (Eq. 4.7) [17]. NMR spectroscopic investigations have found that formation of p-complexes at the double bond adjacent to the carbonyl group, similar to those observed in 1,6-cuprate additions to acceptor-substituted enynes (Sect. 4.2.3), are involved in these transformations. Nevertheless, deeper insight into mechanistic features, which should be highly rewarding for preparative applications, is still awaited.
ð4:7Þ
4.2.2
Acceptor-substituted Enynes
As for conjugate addition reactions of carbon nucleophiles to activated dienes, organocopper compounds represent the reagents of choice for regioselective and stereoselective Michael additions to acceptor-substituted enynes. Whereas substrates bearing an acceptor-substituted triple bond in conjugation with one or even more double bonds (such as 20) react with organocuprates exclusively by 1,4-addition (Eq. 4.8) [18], the corresponding additions to enynes bearing acceptor substituents at the double bond can result in the formation of several regioisomeric products [3o, 8, 19].
ð4:8Þ
Analogously to the acceptor-substituted dienes (Scheme 4.1), the outcome of the reaction depends strongly on the regioselectivity of both the nucleophilic attack of the copper reagent (1,4- or 1,6-addition) and of the electrophilic trapping of the enolate formed (Scheme 4.2). Since the allenyl enolate formed by 1,6-addition can furnish either an allene or a conjugated diene upon reaction with a soft electrophile, and so o ers the possibility of creating axial chirality, this transformation is of special interest from the preparative and also the mechanistic points of view. Recent investigations have demonstrated that the regioselectivities and stereoselectivities of both steps can be controlled by the choice of the reactants, in particular by ‘‘fine-tuning’’ of the organocopper reagent and the electrophile.
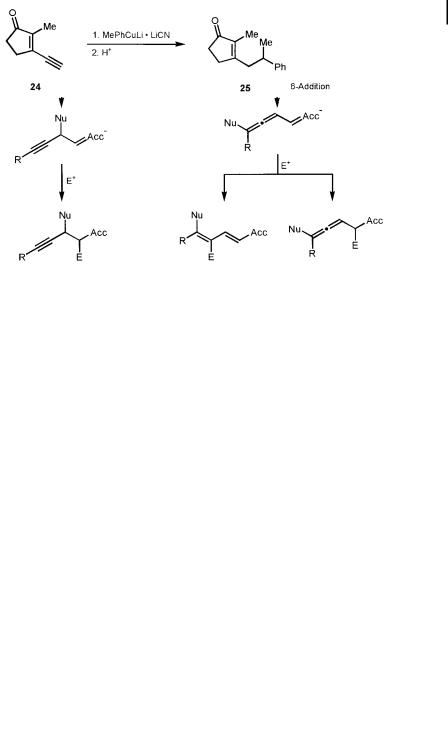
4.2 Copper-mediated Addition Reactions to Extended Michael Acceptors 151
Scheme 4.2. Regioselectivity in conjugate addition reactions to acceptor-substituted enynes.
The first copper-mediated addition reactions to enynes with an acceptor group at the triple bond were reported by Hulce [19, 20], who found that 3-alkynyl-2-cyclo- alkenones 22 react regioselectively with cuprates, in a 1,6-addition at the triple bond (Eq. 4.9). The allenyl enolates thus formed are protonated at C-4 to provide conjugated dienones 23 as mixtures of E and Z isomers. Interestingly, substrates of this type can also undergo tandem 1,6- and 5,6-additions, indicating that the allenyl enolate is su ciently nucleophilic to react with another organometallic reagent in a carbometalation of the allenic double bond distal to the electron-releasing enolate moiety (Eq. 4.10) [20b]. Hence, it is even possible to introduce two di erent groups at the Michael acceptor, either by successive use of two organocopper reagents or by employing a mixed cuprate.
ð4:9Þ
ð4:10Þ

152 4 Copper-mediated Addition and Substitution Reactions of Extended Multiple Bond Systems
With regard to preparative applications, however, shifting the regioselectivity of the electrophilic quenching reaction towards the formation of allenes would be far more interesting, since the scope of synthetic methods for the preparation of functionalized allenes has hitherto been rather limited [21]. Moreover, a stereoselective reaction of this type would open up a route to these axially chiral compounds in enantiomerically enriched or even pure form. The Gilman cuprate Me2CuLi LiI and cyano-Gilman reagents R2CuLi LiCN (R 0Me) in diethyl ether did indeed react regioselectively in a 1,6-fashion with various substituted 2-en-4-ynoates 26. After protonation with dilute sulfuric acid, the b-allenic esters 27, with alkyl, alkenyl, aryl, and silyl substituents, were obtained in good chemical yields (Eq. 4.11) [22].
ð4:11Þ
The nature of the acceptor substituent exerts hardly any influence on the regioselectivity of the cuprate addition to acceptor-substituted enynes. Enynes 28, variously incorporating thioester, lactone, dioxanone, keto, sulfonyl, sulfinyl, cyano, and oxazolidino groups, all react in a 1,6-manner to furnish functionalized allenes 29 (Eq. 4.12). In contrast, though, 1-nitro-l-en-3-ynes are attacked at the CbC double bond, with formation of the corresponding 1,4-adducts [22c]. The di erences in reactivity can be described qualitatively by the following reactivity scale: Acceptor (Acc) ¼ NO2 > COR, CO2R, COSR > CN, SO3R, oxazolidino > SO2R > SOR gCONR2. Remarkably, the regioselectivity of the cuprate addition to acceptor-substituted enynes is also insensitive to the steric properties of the substrate. Thus, enynes with t-butyl substituents at the triple bond (e.g., 30) undergo 1,6-additions even when the cuprate itself is sterically demanding (Eq. 4.13) [22b]. This method is therefore highly useful for the preparation of sterically encumbered allenes of type
31.
ð4:12Þ

4.2 Copper-mediated Addition Reactions to Extended Michael Acceptors 153
ð4:13Þ
In order to achieve acceptable chemical yields with less reactive Michael acceptors, such as sulfones and sulfoxides, it is often necessary to use more reactive organocopper reagents or to activate the substrate by Lewis acid catalysis. Thus, treatment of enyne sulfone 32 with five equivalents of the Gilman cuprate Me2CuLi alone gave no trace of the addition product, whereas the analogous reaction with Me3CuLi2 provided the desired allene 33 only in a disappointing 16% yield (Eq. 4.14) [22c]. With two equivalents of Me2CuLi in the presence of one equivalent of Me3Sil, however, the yield was increased to 45%, although with Me3SiOTf as additive the allene 33 was isolated in only 29% yield. Unfortunately, enyne amides completely fail to form 1,6-adducts even under these conditions.
ð4:14Þ
Unlike the substrate, the organocuprate component has a pronounced influence on the regiochemical course of the addition to acceptor-substituted enynes. While the Gilman cuprate Me2CuLi LiI as well as cyano-Gilman reagents R2CuLi LiCN (R 0Me) readily undergo 1,6-additions, the Yamamoto reagents RCu BF3 [3f ] and organocopper compounds RCu activated by Me3SiI [23] both a ord 1,4-adducts [3o]. In some cases, even 1,4- and 1,6-reduction products are observed; these may be the result of electron transfer from the cuprate to the substrate or of hydrolysis of a stable copper(III) intermediate [19, 24]. Lower order cyanocuprates RCu(CN)Li again show a di erent behavior; although these do not usually react with acceptorsubstituted enynes, the cuprate tBuCu(CN)Li nevertheless undergoes anti-Michael additions with 2-en-4-ynoates and nitriles (Eq. 4.15) [25]. A satisfactory interpretation of the capricious behavior of organocuprates in these conjugate addition reactions to acceptor-substituted enynes is unfortunately still awaited, and so identification of the appropriate reaction conditions for each cuprate often has to rely upon a ‘‘trial and error’’ search.
ð4:15Þ

154 4 Copper-mediated Addition and Substitution Reactions of Extended Multiple Bond Systems
Like the copper-catalyzed 1,4-Michael additions of Grignard reagents to enones and activated dienes, the corresponding 1,6-additions to acceptor-substituted enynes can also be conducted catalytically. However, only very carefully controlled reaction conditions furnish the 1,6-adduct as the major product. Hence, the use of copper (2-dimethylaminomethyl)thiophenolate (18) as catalyst and simultaneous addition of the substrate (e.g., 34) and an organolithium reagent to a suspension of the catalyst 18 in diethyl ether at 0 C resulted in the formation of various substituted b- allenylcarboxylates 36 (Eq. 4.16) [26]. The yields were comparable to those obtained in analogous stoichiometric procedures, whereas only low yields of the 1,6-addition products were found if other copper(I) salts were employed as catalyst, or other Grignard reagents as nucleophile.
ð4:16Þ
As is implicit in the fact that the products of the (stoichiometric) 1,6-cuprate addition
– the lithium allenyl enolate and the organocopper compound – are formed as independent species, it is also possible to conduct the reaction catalytically through in situ regeneration of the cuprate. The reaction can thus be run in a continuous mode, with only catalytic amounts of the preformed cuprate being necessary (with simultaneous addition of the substrate and the organolithium compound) enabling the desired allenes to be prepared even on larger scales (Eq. 4.17) [3o].
ð4:17Þ
As previously mentioned, allenes can only be obtained by 1,6-addition to acceptorsubstituted enynes when the intermediate allenyl enolate reacts regioselectively with an electrophile at C-2 (or at the enolate oxygen atom to give an allenyl ketene acetal; see Scheme 4.2). The regioselectivity of the simplest trapping reaction, the protonation, depends on the steric and electronic properties of the substrate, as well as the proton source. Whereas the allenyl enolates obtained from alkynyl enones 22 always provide conjugated dienones 23 by protonation at C-4 (possibly

4.2 Copper-mediated Addition Reactions to Extended Michael Acceptors 155
through allenyl enols; see Eq. 4.9) [19, 20], the corresponding ester enolates are usually protonated at C-2 (Eq. 4.11), especially if sterically demanding groups at C-5 block the attack of a proton at C-4 (Eq. 4.13) [3o, 22]. In the presence of a substituent at C-2 of the enolate, however, mixtures of both allenes and conjugated dienes are formed for steric reasons (Eq. 4.18). Nevertheless, this problem can be solved by using weak organic acids as a proton source. In particular, pivalic acid (2,2-dimethylpropionic acid) at low temperatures gives rise to exclusive formation of allenes [22a].
ð4:18Þ
In contrast to the protonation, the regioselectivity of reactions between other electrophiles and allenyl enolates derived from 2-en-4-ynoates is independent of the steric and electronic properties of the reaction partners (Scheme 4.3) [3o, 27]. As expected according to the HSAB principle, hard electrophiles such as silyl halides and triflates react at the enolate oxygen atom to form allenyl ketene acetals, while soft electrophiles such as carbonyl compounds attack at C-2. Only allylic and propargylic halides react regioselectively at C-4 of the allenyl enolate to give substituted conjugated dienes. Again, cyclic allenyl enolates obtained through 1,6- cuprate addition to 3-alkynyl-2-cycloalkenones 22 show a deviant behavior; treatment with iodomethane gave product mixtures derived from attack of the electrophile at C-2 and C-4, while the reaction with aldehydes and silyl halides took place exclusively at C-4 [19, 28].
Scheme 4.3. Regioselectivity of trapping reactions of acyclic allenyl enolates with di erent electrophiles.
Several preparative applications of the 1,6-cuprate addition to acceptor-substituted enynes have been described in recent years. In addition to its use in the formation of

156 4 Copper-mediated Addition and Substitution Reactions of Extended Multiple Bond Systems
sterically encumbered allenes (Eq. 4.13) [22b] and simple terpenes such as pseudoionone [22a], this method is also the synthesis of valuable for access to allenic natural products (Eq. 4.19) [3o]. For example, 1,6-addition of lithium di-n-octyl- cuprate to enynoate 40, followed by regioselective protonation with pivalic acid, yielded allene 41, which was then readily convertible into the insect pheromone methyl 2,4,5-tetradecatrienoate (42). Further applications of 1,6-additions in natural product synthesis rely upon vinylallenes as diene components in the Diels–Alder reactions (Eq. 4.20). Hence, the synthesis of the fungal metabolite (G)-sterpurene (46) and some oxygenated metabolites started with the 1,6-addition of lithium dimethylcuprate to enynoate 43 and subsequent regioselective enolate trapping with methyl triflate [29]. The vinylallene 44 thus formed underwent an intramolecular ½4þ2& cycloaddition to give the tricyclic product 45, which was finally converted into the target molecule 46.
ð4:19Þ
ð4:20Þ
The Diels–Alder reaction outlined above is a typical example of the way in which axially chiral allenes, accessible through 1,6-addition, can be utilized to generate new stereogenic centers in a selective fashion. This transfer of chirality is also possible by means of intermolecular Diels–Alder reactions of vinylallenes [30], aldol reactions of allenyl enolates [31], and Ireland–Claisen rearrangements of silyl allenylketene acetals [32].

4.2 Copper-mediated Addition Reactions to Extended Michael Acceptors 157
Recently, the oxidation of titanium allenyl enolates (formed by deprotonation of b-allenylcarboxylates of type 36 and transmetalation with titanocene dichloride) with dimethyl dioxirane (DMDO) was found to proceed regioselectively at C-2. In this way, depending on the steric demand of the substituents at the allenic moiety, the corresponding 2-hydroxy-3,4-dienoates were obtained diastereoselectively with up to 90% ds (Eq. 4.21) [33]. a-Hydroxyallenes of this type are synthetically valuable precursors for 2,5-dihydrofurans, found not only in several natural products but also in biologically active compounds [34]. Thus, the cyclization of allene 47 to heterocycle 48 took place with complete axis-to-center chirality transfer, being easily achieved by treatment with HCl gas in chloroform, acidic ion exchange resins such as Amberlyst 15, or, last but not least, with catalytic amounts of gold(III) chloride (this last method is particularly useful for a-hydroxyallenes containing acid-sensi- tive groups [33b]).
ð4:21Þ
Allenic amino acid derivatives 50, which are of special interest as selective vitamin B6 decarboxylase inhibitors [35], are accessible through 1,6-cuprate addition to 2- amino-substituted enynes 49 (Eq. 4.22) [36]. Because of the low reactivity of these Michael acceptors, however, the reaction succeeds only with the most reactive cuprate: the t-butyl cyano-Gilman reagent tBu2CuLi LiCN. Nevertheless, the addition products are obtained with good chemical yields, and selective deprotection of either the ester or the amino functionality under acidic conditions provides the desired target molecules.
ð4:22Þ
By starting with enantiomerically enriched or pure b-allenylcarboxylates, it is possible to carry out several of the transformations mentioned above stereoselectively. With regard to the required substrates, chiral 5-alkynylidene-1,3-dioxan-4-ones of