

14. S-Nitroso compounds, formation, reactions and biological activity |
675 |
and is believed to be at the diffusion-controlled limit for reaction of NOC (or H2NO2C ) with thiourea54. Alkyl thioureas react with much the same rate constant.
+ |
|
(NH2)2CS C HNO2 C HC D (NH2)2CSNO C H2O |
(25) |
Rate D k[(NH2)2CS][HNO2][HC ] |
(26) |
As expected, other carriers of NOC such as nitrosamines55, alkyl nitrites56 and a nitrososulphonamide57 will also generate the S-nitrososulphonium ion from thiourea.
There appears to be little reported work on S-nitrosation reactions of simple thioketones. Thiocamphor when treated with iso-amyl nitrite in fact gives the oxime58 (formerly called ˛ isonitroso compounds), presumably via the tautomeric form of the thione, i.e. the enethiol. In this respect the reaction is very similar to the reactions of ketones59 which give oximes or C-nitroso compounds via the enol intermediates60.
B. Reactions
The S-nitrososulphonium ions derived from thiourea and its derivatives are not stable in solution but decompose to give the disulphide cation (C,C-dithiodiformamidinium) which has been isolated as its salts, e.g. the perchlorate and characterized by X-ray crystal analysis61 (equation 27). This is an example of the more general reaction whereby, eventually, thioureas and also thioketones and thiocarbonates can be converted to the stable -S-S-dication by a range of oxidizing agents both chemical and electrochemical62.
+ |
+ + |
|
2(NH2)2CSNOD(NH2)2CSSC(NH2)2 C 2NO |
27 |
|
The decomposition reaction has been studied mechanistically63 and the kinetics have
been interpreted in terms of the parallel pathways, one involving a bimolecular reaction
+
between two (NH2)2CSNO species (leading to a second-order kinetic term) and the other a reversible formation of a radical intermediate [(NH2)2CSSC(NH2)2]Cž .
At higher acidities the S-nitrosation reaction of thiourea leads to the formation of urea64 (equation 28) via, it is believed, the intermediate formation of the S-nitroso species. The reaction can also be brought about by nitrosamines or alkyl nitrites as the carriers of NOC . Reaction is thought to involve nucleophilic attack of the intermediate by water or the elimination of HSNO giving a carbodiimide, which is then hydrated.
C S NO+
+
C S NO H2O
C O |
(28) |
An important property of the S-nitroso thiourea derivatives is the ability to effect electrophilic nitrosation of any of the conventional nucleophilic centres. This is manifest kinetically by the catalysis of nitrous acid nitrosation effected by added thiourea (equation 29). The situation is completely analogous to the catalysis of the same reactions by added halide ion or thiocyanate ion. The catalytic efficiency of thiourea depends on both the equilibrium constant KXNO for the formation of the intermediate and also its rate constant k with typ-
ically a secondary amine65. Since KXNO is known (5000 dm6 mol 2), it is easy to obtain
+
values of k. Values of k follow the trend (NH2)2CSNO < ONSCN < BrNO < ClNO whereas KXNO values follow the opposite trend. Since the range of KXNO values is very much larger than that of K values, the most efficient catalyst by far is thiourea. This is illustrated graphically in Figure 2 where the catalytic efficiencies of thiourea, thiocyanate
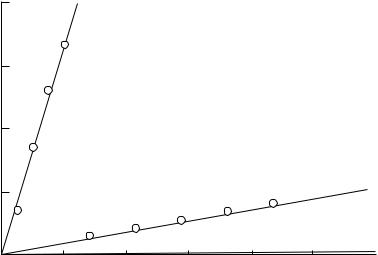
676 |
D. Lyn H. Williams |
104 k0 (s−1)
400
SC(NH2)2
Nitrosation of morpholine
300
200
SCN−
100
Br−
0 |
5 |
10 |
15 |
20 |
25 |
|
103 [X− ] (mol dm−3)
FIGURE 2. Catalysis by Br , SCN and (NH2)2CS in the nitrosation of morpholine
ion and bromide ion are compared in the nitrosation of morpholine by nitrous acid66. A similar analysis has been carried out for the diazotisation of aniline67 and earlier for aliphatic amines68. It appears that thiourea is the most efficient catalyst of nitrosation using aqueous nitrous acid.
HNO2 |
|
HC |
|
(NH2)2CS |
KXNO |
+ |
|
H2O |
C |
C |
! |
(NH2)2CSNO |
C |
||||
|
|
|
|
|
29 |
|||
|
|
+ |
|
|
k |
|
|
|
|
|
|
|
|
|
|
||
(NH2)2CSNO C PhN(Me)H |
! PhN(Me)NO |
|
|
|||||
IV. FORMATION AND REACTIONS OF OTHER S-NITROSO COMPOUNDS
A. S-Nitrososulphides
It has been known for a long time that simple alkyl sulphides yield coloured (yellow or red) solutions when treated with alkyl nitrites or nitrous acid69. The colours fade on standing and products have not been identified. It does, however, seem likely that the coloured products are in fact S-nitroso ions (equation 30) by analogy with the corresponding reaction of thiols, although in this case there is no suitable leaving group which would lead to a stable product.
+ |
|
R2S C ‘NOC ’ D R2 SNO |
30 |
There is indirect kinetic evidence that S-nitrosation of a sulphide occurs70, followed by a S to N rearrangement of the nitroso group, leading finally to deamination (equation 31). The evidence is based on the much higher reaction rate when the sulphur atom is present.
14. S-Nitroso compounds, formation, reactions and biological activity |
677 |
S to N transfer of the nitroso group has also been postulated on a number |
of other |
occasions53,71 to explain enhanced reaction rates. Detailed kinetic studies on the nitrosation of thioproline72 (12) and thiomorpholine73 (13) reveal that two pathways can exist, which depend on the reaction conditions, (a) a direct reaction at nitrogen and (b) a direct reaction at sulphur followed by S to N migration of the nitroso group.
CH2SMe
HNO2
C
NH2
+ |
Me |
|
CH2SMe |
CH2SMe |
|
CH2S |
|
|
|||
NO |
|
|
|
|
|
C |
C |
+ |
|
C |
|
|
|||||
NH2 |
|
|
|
OH |
|
|
|
NH2NO |
|||
31
S
S
N |
CO2H |
N |
H |
|
H |
(12) |
(13) |
|
Sulphides should be capable of bringing about catalysis of nitrosation if the |
+ |
|
S−NO |
||
ion is formed and if it is itself an electrophilic nitrosating agent. This has been shown to be the case74 for the nitrosation of N-methylaniline in the presence of added dimethyl
sulphide. The overall catalytic effect is approximately the same as that shown by bromide
+
S−NO
formation or for the rate constant for its reaction with the amine. Diazotisation of aniline is also catalysed by S-methylcysteine66. Evidence that S-nitroso sulphides can be generated from nitrosamines and sulphides comes from the substantial nucleophilic reactivity of methionine (approximately the same level as bromide ion) in the denitrosation of an aromatic nitrosamine in acid solution in the presence of a ‘nitrite trap’ such as hydrazine75.
B. S-Nitrososulphinates (Sulphonyl Nitrites)
S-Nitrososulphinates can be made by treating sulphinic acids with N2O4 at about20 °C in ether (equation 32)76. Use of nitrous acid on alkyl nitrites leads to the formation of the corresponding hydroxylamines (equation 33) in a reaction where it is believed that the first formed nitrososulphinate nitrosates another molecule of the reactant sulphinic acid77.
|
|
|
Et2O |
|
|
RSO2H |
C |
N2O4 |
! |
RSO2NO |
(32) |
|
|
|
|
|
|
|
|
|
20 °C |
|
|
RSO2H C HNO2 D RSO2NO |
(33) |
||||
RSO2NO C RSO2H D (RSO2)2NOH |
|
||||
The isolated nitrososulphinates are unstable brown crystals with the NDO IR absorption band near 1840 cm 1, i.e. at shorter wavelengths than in nitroso thiols (1490 1700 cm 1) due to the powerful electron-withdrawing effect of the sulphonyl group. They decompose
678 |
D. Lyn H. Williams |
upon warming, giving off nitric oxide and forming the hydroxylamine derivative. They are claimed to be the most powerful nitrosating agents known, although there are no quantitative data available. They react with alcohols giving alkyl nitrites, thiols to give S- nitrosothiols and provide excellent reagents for deamination of aryl amines in acetonitrile containing anhydrous Cu(II) halides at room temperature78. Nitrosamines are formed from secondary amines in high yield at room temperature11. The most used reagent to date is 4-MeC6H4SO2NO.
The kinetics of the nitrosation of benzenesulphinic acid have been determined79. The reaction is very rapid and requires stopped-flow techniques. This makes benzenesulphinic acid an excellent trap for free nitrous acid, on par with the more well-known hydrazoic acid and hydrazinium ion80. In mildly acid solution reaction occurs via the sulphinic acid molecule and also the sulphinate ion. As expected, the latter is the more reactive and reaction takes place at the diffusion limit. All evidence points to the fact that the first nitrosation by NOC is the rate-limiting step.
C. S-Nitroso Compounds with Inorganic Anions
1. Nitrosyl thiocyanate
When thiocyanate ions are added to nitrous acid in water, a pink colouration develops which is believed to be due to the formation of nitrosyl thiocyanate (equation 34), which is too unstable to be isolated but which can be used as a nitrosating agent in aqueous solution. Because the equilibrium constant for ONSCN formation81 is quite large (30 dm6 mol 2) at 25 °C, thiocyanate ion is an excellent catalyst for aqueous electrophilic nitrosation. The well established82 series is Cl < Br < SCN < (NH2)2CS. Thiocyanate ion is also a sufficiently powerful nucleophile to react in acid solution with nitrosamines in a denitrosation process (equation 35), which can only be driven to the right if the nitrosyl thiocyanate is removed by, e.g., reaction with a ‘nitrite trap’ such as hydrazoic acid.
HNO2 |
C |
HC |
C |
SCN |
! |
ONSCN |
C |
H2O |
(34) |
+ |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
(35) |
|
PhN(Me)HNO C SCN ! PhNMeH C ONSCN |
|||||||||
Removed
There are numerous examples of the demonstration of the catalytic activity of thiocyanate ion for a wide variety of substrates. In general the reaction of ONSCN is rate-limiting, but in some cases (just as for the nitrosyl halides) with very reactive substrates the formation of ONSCN can be rate-limiting.
Although ONSCN has not been isolated, ab initio calculations83 and application of HASB theory suggest that the nitroso group is bonded to the sulphur atom.
2. Nitrosyl thiosulphate
A yellow solution is formed when nitrous acid is added to thiosulphate ion in water84. This is believed to be due to the formation of nitrosyl thiosulphate [O3SSNO] , although this has not been isolated and even in solution decomposition is fairly rapid. The equilib-
rium constant for its formation KXNO is 1.66ð107 dm6 mol 2 at 25 °C and the UV-visible absorption spectrum is very similar to that of other S-nitroso compounds85. The rate constant for its formation is very large and is believed to represent a diffusion controlled process. Thiosulphate ion does appear to catalyse nitrosation but, over the range studied
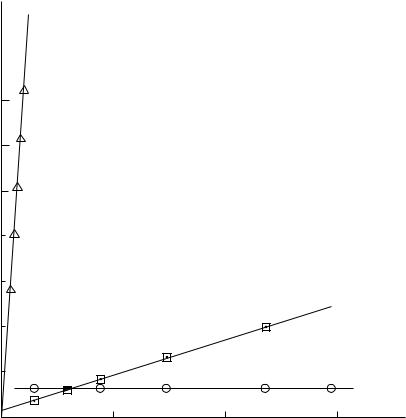
14. S-Nitroso compounds, formation, reactions and biological activity |
679 |
102k0 (s−1)
SCN−
7
6
5
4
3
Br−
2 |
|
|
|
1 |
|
|
S2O32− |
0 0 |
1 |
2 |
3 |
103[Nucleophile] (mol dm−3)
FIGURE 3. Catalytic effect of added Br , SCN and S2O32 in the nitrosation of N-methylaniline
for the reaction of N-methylaniline, there is no kinetic dependence of the rate constant
upon [S2O32 ] (see Figure 3)86. This is because KXNO is so large that under the experimental conditions HNO2 is fully converted to O3SSNO . A kinetic analysis reveals that
O3SSNO is not a very effective nitrosating agent, being several orders of magnitude less reactive than ONSCN.
3. Nitrosyl bisulphite
It has been known since 1894 that bisulphite ion reacts with nitrous acid to give hydroxylamine disulphonate87 (equation 36). It is believed that the nitrosyl bisulphite species is an intermediate which then nitrosates a further bisulphite ion. In this regard the reaction bears a striking resemblance to the nitrosation of sulphinic acids discussed earlier. This reaction has been important commercially for some time as the Raschig process88 for the production of hydroxylamine, since the disulphonate is readily hydrolysed to give the
680 |
D. Lyn H. Williams |
free hydroxylamine. The results suggest that HO3SNO is indeed a very reactive nitrosating species (just as the S-nitrososulphinates, see Section IV.B) but no kinetic studies have been reported on the possible catalytic activity of bisulphite ion in nitrosation reactions.
NOC C HSO3 D HO3SNO
HC |
|
HO3SNO C HSO3 ! [(HO3S)2NO] ! (HO3S)2NOH |
(36) |
V.REFERENCES
1.D. L. H. Williams, Chem. Soc. Rev., 14, 171 (1985).
2.L. R. Dix and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 109 (1984); P. A. Morris and
D.L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 513 (1988).
3.H. Lecher and W. Siefhen, Chem. Ber., 59, 1314, 2594 (1926).
4.H. S. Tasker and H. O. Jones, J. Chem. Soc., 95, 1910 (1909).
5.R. J. Phillips and H. Moor, Spectrochim. Acta, 17, 1004 (1961); R.J. Phillips, J. Mol. Spectrosc., 6, 492 (1961).
6.S. Oae, Y. H. Kim, D. Fukushima and K. Shinhama, J. Chem. Soc., Perkin Trans. 1, 913 (1978).
7.J. Mason, J. Chem. Soc. (A), 1587 (1969).
8.M. P. Doyle, J. W. Terpstra, R. A. Pickering and D. M. LePoire, J. Org. Chem., 48, 3379 (1983);
S.A. Glover, A. Goosen, C. W. McClelland and F. R. Vogel, S. Afr. J. Chem., 34, 96 (1981).
9.H. M. S. Patel and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 37 (1990).
10.S. M. N. Y. F. Oh and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 755 (1989).
11.S. Oae and K. Shinhama, Org. Prep. Proced. Int., 15, 165 (1983).
12.H. Rheinboldt, Chem. Ber., 59, 1311 (1926).
13.G. Kresze and U. Uhlich, Chem. Ber., 92, 1048 (1959).
14.L. Field, R. V. Dilts, R. Ravichandran, P. G. Lenhert and G. E. Carnahan, J. Chem. Soc., Chem. Commun., 249 (1978).
15.T. W. Hart, Tetrahedron Lett., 26, 2013 (1985).
16.B. Roy, A. du M. d’Hardemare and M. Fontecave, J. Org. Chem., 59, 7019 (1994).
17.H. A. Moynihan and S. M. Roberts, J. Chem. Soc., Perkin Trans. 1, 797 (1994).
18.J. Barrett, D. F. Debenham and J. Glauser, J. Chem. Soc., Chem. Commun., 248 (1965).
19.S. Moncada, R. M. J. Palmer and E. A. Higgs, Pharmacol. Rev., 43, 109 (1991).
20.A. R. Butler and D. L. H. Williams, Chem. Soc. Rev., 233 (1993).
21.P. L. Feldman, O. W. Griffith and D. J. Stuehr, Chem. Eng. News, 20, 26 (1993).
22.M. Fontecave and J -L. Pierre, Bull. Soc. Chim. Fr., 131, 620 (1994).
23.J. Barrett, L. J. Fitygibbones, J. Glauser, R. H. Still and P. N. W. Young, Nature, 211, 848 (1966).
24.H. Rheinbolt and F. Mott, J. Prakt. Chem., 133, 328 (1932).
25.D. A. Wink, J. F. Darbyshire, R. W. Nims, J. E. Saavedra and P. C. Ford, Chem. Res. Toxicol., 6, 23 (1993).
26.S. C. Askew, D. J. Barnett, J. McAninly and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 741 (1995).
27.J. McAninly, D. L. H. Williams, S. C. Askew, A. R. Butler and C. Russell, J. Chem. Soc., Chem. Commun., 1758 (1993).
28.I. M. Klotz, G. H. Czerlinski and H. A. Friess, J. Am. Chem. Soc., 80, 2920 (1958).
29.F. J. Davis, B. C. Gilbert, R. O. C. Norman and M. C. R. Symons, J. Chem. Soc., Perkin Trans. 2, 1763 (1983).
30.B. Saville, Analyst, 83, 670 (1958).
31.H. R. Swift and D. L. H. Williams, J. Chem. Soc. Perkin Trans. 2, (1996) to appear.
32.S. S. Al-Kaabi, D. L. H. Williams, R. Bonnett and S. L. Ooi, J. Chem. Soc., Perkin Trans. 2, 227 (1982).
33.H. Rheinboldt and F. Mott, Chem. Ber., 65, 1223 (1932).
34.Y. H. Kim, K. Shinhama, D. Fukushima and S. Oae, Tetrahedron Lett., 1211 (1978).
35.J. W. Park, Biochem. Biophys. Res. Commun., 152, 916 (1988).
36.D. J. Meyer, H. Kramer, N. Ozer, B. Coles and B. Ketterer, FEBS Lett., 345, 177 (1994).
37.D. J. Barnett, A. Rios and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1279 (1995).

14. S-Nitroso compounds, formation, reactions and biological activity |
681 |
38.W. R. Mathews and S. W. Kerr, J. Pharmacol. Exp. Ther., 267, 1529 (1993) and references cited therein.
39. J. Loscalzo, J. Clin. Invest., 76, 703 (1985); M. W. Radomski, D. D. Rees, A.Dutra and
S.Moncada, Br. J. Pharmacol., 107, 745 (1992).
40.E. J. Langford, A. S. Brown, R. J. Wainwright, A. J. de Belder, M. R. Thomas, R. E. A. Smith,
M.W. Radomski, J. F. Martin and S. Moncada, Lancet, 344, 1458 (1994).
41.A. de Belder, C. Lees, J. Martin, S. Moncada and S. Campbell, Lancet, 345, 124 (1995).
42. L. J. Ignarro, H. Lippton, J. C. Edwards, W. H. Baricos, A. L. Hyman, P. J. Kadowitz and
C.A. Gruetter, J. Pharmacol. Exp. Ther., 218, 739 (1981); G. M. Rubanyi, A. Johns, D. Wilcox,
F.N. Bates and D. Harrison, J. Cardiovasc. Pharmacol., 17, S41 (1991).
43.J. S. Stamler, O. Jaraki, J. Osborne, D. I. Simon, J. Keaney, J. Vita, D. Singel, C. R. Valeri and
J.Loscalzo, Proc. Natl. Acad. Sci. U.S.A., 89, 7674 (1992).
44.B. Gaston, J. Reilly, J. M. Drazen, J. Fackler, P. Ramdev, D. Arnelle, M. E. Mullins, D. J. Sugarbarker, C. Chee, D. J. Singel, J. Loscalzo and J. S. Stamler, Proc. Natl. Acad. Sci. U.S.A., 90, 10957 (1993).
45.D. I. Simon, J. S. Stamler, O. Jaraki, J. F. Keaney, J. A. Osborne, S. A. Francis, D. J. Singel and
J.Loscalzo, Arteriosclerosis Thrombos., 13, 791 (1993).
46.P. R. Myers, R. L. Minor, R. Guerra, J. N. Bates and D. G. Harrison, Nature, 345, 161 (1990).
47.J. A. Bauer and H -L. Fung, J. Pharmacol. Exp. Ther., 256, 249 (1990).
48.E. A. Kowaluk and H. Fung, J. Pharmacol. Exp. Ther., 256, 1256 (1990).
49.D. R. Arnelle and J. S. Stamler, Arch. Biochem. Biophys., 318, 279 (1995).
50.M. P. Gordge, D. J. Meyer, J. Hothersall, G. H. Neild, N. N. Payne and A. Noronha-Dutra, Br.
J.Pharmacol., 114, 1083 (1995).
51.R. G. Pearson, H. Sobel and J. Songstad, J. Am. Chem. Soc., 90, 319 (1968).
52.A. E. Werner, J. Chem. Soc., 101, 2180 (1912); M. E. Coade and A. E. Werner, J. Chem. Soc., 102, 1221 (1913).
53.K. Al-Mallah, P. Collings and G. Stedman, J. Chem. Soc., Dalton Trans., 2469 (1974).
54.P. Collings, K. Al-Mallah and G. Stedman, J. Chem. Soc., Perkin Trans. 2, 1734 (1975).
55.D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 128 (1977).
56.M. J. Crookes and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 759 (1989).
57.J. R. Leis, M. E. Pena˜ and A. M. Rios, J. Chem. Soc., Perkin Trans. 2, 587 (1995).
58.D. C. Sen, J. Indian Chem. Soc., 12, 751 (1935).
59.O. Touster, in Organic Reactions (Ed. R. Adams), 7, Chap. 6, Wiley, New York, 1953, p. 327.
60.J. R. Leis, M. E. Pena,˜ D. L. H. Williams and S. D. Mawson, J. Chem. Soc., Perkin Trans. 2, 157 (1988).
61.O. Foss, J. Johnsen and O. Tvedten, Acta Chem. Scand., 12, 1782 (1958).
62.R. L. Blankespoor, M. P. Doyle, D. M. Hedstrand, W. H. Tamblyn and D. A. Van Dyke, J. Am. Chem. Soc., 103, 7096 (1981).
63.P. Collings, M. Garley and G. Stedman, J. Chem. Soc., Dalton Trans., 331 (1981).
64. K. A. Jorgensen and S. O. Lawesson, Chem. Ser., 20, 227 (1982); J. W. Lown and
S.M. S. Chauhan, J. Org. Chem., 48, 507 (1983).
65.D. L. H. Williams, Nitrosation, Cambridge University Press, 1988, pp. 22 24 and references cited therein.
66.T. A. Meyer and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 361 (1981).
67.L. R. Dix and D. L. H. Williams, J. Chem. Res. (S), 97 (1984).
68.M. Masui, C. Ueda, T. Yasuoka and H. Ohmori, Chem. Pharm. Bull., 27, 1274 (1979).
69.E. M. Harper and A. K. Macbeth, Proc. Chem. Soc., 30, 15 (1914); A. K. Macbeth and D. D. Pratt,
J.Chem. Soc., 119, 354 (1921).
70.T. A. Meyer and D. L. H. Williams, J. Chem. Soc., Chem. Commun., 1067 (1983).
71. J. W. Lown and S. M. S. Chauhan, J. Org. Chem., 48, 3901 (1983); T. Tahira, M. Tsuda,
K. Wakabayashi, M. Nago and T. Sugimura, Gann, 75, 889 (1984).
72.A. Castro, E. Iglesias, J. R. Leis, J. V. Tato, F. Meijide and M. E. Pena,˜ J. Chem. Soc., Perkin Trans. 2, 651 (1987).
73.A. Coello, F. Meijide and J. V. Tato, J. Chem. Soc., Perkin Trans. 2, 1677 (1989).
74.T. Bryant and D. L. H. Williams, J. Chem. Res. (S), 174 (1987).
75.G. Hallett and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 624 (1980).
76.S. Oae, K. Shinhama and Y. H. Kim, Tetrahedron Lett., 3307 (1979).
77.C. S. Marvel and R. S. Johnson, J. Org. Chem., 13, 822 (1948); G. Kresze and W. Kort, Chem. Ber., 94, 2624 (1961).

682 |
D. Lyn H. Williams |
78.S. Oae, K. Shinhama and Y. H. Kim, Bull. Chem. Soc. Jpn., 53, 1065 (1980).
79.T. Bryant and D. L. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1083 (1985).
80.G. P. Quinton and D. L. H. Williams, J. Chem. Res. (S), 209 (1987).
81.G. Stedman and P. A. E. Whincup, J. Chem. Soc., 5796 (1963).
82.Reference 65, pp. 10 24.
83.K. A. Jorgensen and S. O. Lawesson, J. Am. Chem. Soc., 106, 4687 (1984).
84.J. O. Edwards, Science, 113, 392 (1951).
85.M. S. Garley and G. Stedman, J. Inorg. Nucl. Chem., 43, 2863 (1981).
86.T. Bryant, D. L. H. Williams, M. H. H. Ali and G. Stedman, J. Chem. Soc., Perkin Trans. 2, 193 (1986).
87.E. Divers and T. Haga, J. Chem. Soc., 65, 523 (1894).
88.F. Raschig, Z. Angew. Chem., 17, 1398 (1904).