

2. Mass spectra of double-bonded groups |
63 |
reaction pathways can be recognized, such as ˛-, - and υ-cleavage, simple and double McLafferty rearrangements, Et and Pr loss, cyclizations, etc. The same authors also use these substances to demonstrate the superiority of a pentaquadrupole instrument52. The combination of the different ion manipulations necessary for the reaction intermediate scans hide pitfalls and can produce errors that are not always immediately visible, as is demonstrated with 2-octanone by Eller and Drewello524. It is of interest that in the time window of FIK 7-methyl-octan-4-one-7-D1 a the H/D isomerization competes with even the fastest fragmentation, whereas this is not the case for 2-octanone-1,1,1,3,3-D5 b, as is shown by Derrick and collaborators525,526. Partial H D randomization occurs prior to the McLafferty rearrangement. This behavior contrasts sharply with that of the straight-chain ketone b in which no such randomization occurs prior to the McLafferty rearrangement at times <0.7 ns. At times of 10 100 ps following Þeld ionization (FI) the rate of the McLafferty rearrangement is an order of magnitude smaller in a than in b, but at ca 1 s the situation is reversed with the rate being an order of magnitude smaller in the straightchain ketone. The difference in kinetics is consistent with the McLafferty rearrangement at times > a few 10 11 s following FI being a stepwise rather than a concerted process. It is proposed that H D randomization in a involves -transfer to the oxygen forming a tertiary radical, followed by reverse D transfer from the oxygen to the alkyl chain. The reverse transfer is facilitated by the stability of the tertiary radical.
X. DIELS ALDER AND RETRO-DIELS ALDER REACTIONS
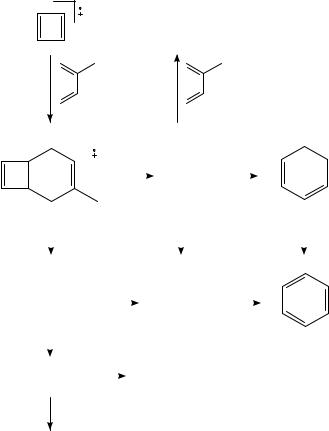
The Diels Alder (DA) reaction is in its simplest form a [4C2] cyclization between a diene and a dienophile. The retro-Diels Alder (RDA) reaction is the opening in the simplest case of a substituted cyclohexene as is shown in Scheme 11. According to Winters and Collins527, the energy required to produce C4H6Cž + C2H4 from [cyclohexene]Cž lies with 169 kJ mol 1 near the thermochemical threshold. Although not many pericyclic reactions of doublet (spin) species are symmetry-allowed528, the DA is such a reaction (for a discussion see elsewhere529,530): The orbital correlation diagram reveals that cycloaddition of the ethylene cation radical to s-cis-1,3-butadiene is symmetry-allowed531. However, the reactions are not limited to six-membered rings532. They are of considerable importance in organic chemistry, especially because of their stereoselectivity. Since they are in addition much faster for cations than for neutrals533, products or fragments of DA or RDA are of analytic interest in mass spectrometry. However, care has to be taken that the reaction does not already take place with the neutral substance in the inlet system, as has been demonstrated e.g. by Veith and Hesse534 for different classes of compounds. It will be shown later that this can also be used for producing new, unstable compounds by pyrolysis.
RDA |
|
DA |
+ |
SCHEME 11
It is not astonishing that a series of reviews covers this interesting Þeld. After a Þrst review by Kuhne and Hesse535 on RDA reactions of tetralin and its derivatives, a ÔclassicalÕ overlook of the Þeld was given in 1984 by Turecekÿ and Hanusÿ529. A chapter in this series by Mruzek25 covers the Þeld up to 1988. The literature in the Þeld of mass

64 |
Tino Gaumann¬ |
spectrometry of large oligonucleides, primarily by electrospray and laser desorption methods, is summarized for the period 1988 through mid-1991 by McCloskey and Crain536. Mandelbaum537 treated in 1993 the stereochemical effects of the RDA fragmentation in a book about the applications of mass spectrometry in organic stereochemistry538. Many cases of RDA fragmentations are cited in the review by Vairamani and Saraswathi73 on a mass spectral study of geometrical E- and Z-isomers. The DA reaction of ÔneutralÕ molecules has been reviewed by Fringuelli and Taticchi539.
Bouchoux and Salpin540 studied the simplest DA reaction in detail: [1,3-butadiene]Cž C ethylene, using also labeled compounds. Because of the low pressure in the ion cyclotron resonance instrument no stable adduct [C6(H/D)10]Cž is seen, only ions corresponding to the loss of Me and ethylene. Measurements of the deprotonation enthalpies indicate that the [C5H7]C ions have the cyclopentadienyl structure. The distribution of the deuterium atoms in the products is nearly random, contrary to the loss of ethylene, where very little scrambling is observed. The ethylene contains one terminal CH2 group of butadiene and one CH2 from ethylene. Since the two fragmentations have about the same time dependence, it can be assumed that they originate from the same adduct, which the authors propose to have the structure of the distonic ion CH2DCHCC HCH2CH2CH2ž . That this reaction occurs without critical energy is concluded by Bauld541 from a MO calculation. This ion either isomerizes to a vinycyclobutene that can split off an ethylene, or it undergoes several hydrogen shifts causing the H/D scrambling and Þnally leading to a Me-cyclopentadiene that loses the methyl group. The complex addition dissociation of 1,3-butadiene cation and ethylene precludes a synchronous character for the DA reaction.
The RDA reaction is a high-energy process that is in competition with other processes that need less activation energy, albeit with higher steric hindrance. Even if one limits the RDA process as an elimination of a dienophile that contains two consecutive carbon atoms from the ring (in the correct initial position), there is still the possibility of a concerted or a two-step process, possibly in competition. In the latter case, the stereospeciÞc characteristic of the RDA fragmentation may be lost. Several reactions yielding the same products may compete with each other, asking for much experimental ingenuity in order to get a clear-cut answer. This may be illustrated by the classic elimination of ethylene from tetralin that had been studied by Budzikiewicz, Levsen and collaborators542,543; see also Mruzek25 (Scheme 12). The time dependence of the reaction is studied with the help of Þeld ionization kinetics, using 13C- and D-labeled compounds. It results that ethylene is not only lost in a RDA reaction from position 3 C 4, but also from 1 C 4 and 2 C 3 and after complete scrambling of all C and D atoms of the saturated ring. The time dependence demonstrates that the high-energy RDA process prevails at short times, giving way after 1 ns to the process that eliminates ethylene from positions 1 C 2 and 3 C 4, which is still the main process in the metastable window (>1 s), although an important scrambling sets in after 1 s. Therefore, it can be concluded that the metastable decomposition and the structure of the molecular ion that is used for the study of fragmentation by collisional activation do not necessarily correspond to the structure of the initial molecular
1
2 |
|
C |
H + |
+ C |
H |
3 |
|
||||
8 |
8 |
2 |
4 |
||
4
SCHEME 12
2. Mass spectra of double-bonded groups |
65 |
ion after ionization. It can be assumed that the resulting C8H8Cž ion consists of a mixture of structures. The RDA fragmentation might yield the o-quinodimethane ion, the 1,2-elimination may yield styrene. CID experiments give a spectrum that is somewhere midway between the corresponding spectra of styrene and the C8H8Cž ion formed from tetrahydroisoquinoline. The latter fragments in a pure RDA reaction. The ideal case for such a study is the ÔsymmetricÕ molecule 5,6,6a,7,12,12a-hexahydrobenzo[a]anthracene, studied by Bobenrieth, Levy and Hass544 and amply discussed by Mruzek25.
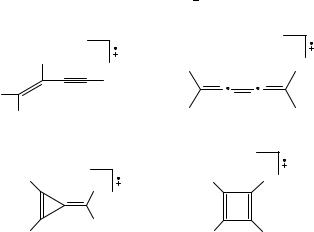
C4H4Cž is an interesting candidate for a study of the DA reaction, since in spite of its simple formula it exists in several isomeric forms that can be prepared separately. Its rate of formation as a function of the internal energy of benzene, one of its precursors, has been measured by Andlauer and Ottinger545,546. Rosenstock and collaborators547,548 propose the existence of linear and cyclic forms. Several attempts to elucidate the structure of C4H4Cž are summarized by Baer and collaborators549 who studied its formation, together with that of C3H3C , as a function of the internal energy of 2,4- and 1,5-hexadiyne as precursors. They suggest the structure of a methylene-cyclopropene cation (c in Scheme 13) for C4H4Cž . Benzene and pyridine as precursors were studied by Ausloos550. In the fragmentations two C4H4Cž are formed, only one of them reacting with benzene. On the basis of CT reactions he attributes the structure of 3-butene-1-yne (a) to the reactive form and methylenecyclopropene (c) to the unreactive form. Bowers and collaborators551 conÞrm the Þndings by Ausloos and show that the linear form can be isomerized with acetylene to the cyclic form over a long-lived C6H6Cž intermediate. Van der Hart137, studying the photofragmentation of benzene and 1,5-hexadiyne, determined that in these fragmentations 60% a, 10% b, and 30% d are formed, measured on the time scale (milliseconds) of an ion cyclotron resonance spectrometer. When studying the ion/molecule reactions, unimolecular dissociations and CID spectra of a series of precursors, the group of Lifshitz and Levsen552 adds cyclobutadiene (d) as an additional structure to the list. Kohn and Chen553 measured the photoelectron spectrum of cyclobutadiene, determined an ionization energy of 8.16 š 0.03 eV and conÞrmed the rectangular structure for the neutral and the ion. The spectrum conÞrms that correlation between and electrons is needed in an ab initio calculation for the ion to obtain even a qualitative correct geometry for the Jahn-Teller distorted ion. The group of McLafferty554 556 conÞrms all four structures with
|
H |
H |
|
H |
|
|
|
||
|
|
H |
|
|
H |
|
|
|
|
H |
|
H |
|
H |
|
(a) |
|
(b) |
|
|
|
|
||
H |
|
H |
H |
H |
|
|
|
|
|
H |
|
H |
H |
H |
|
|
|||
|
|
(c) |
|
(d) |
|
|
SCHEME 13 |
|
|

66 |
Tino Gaumann¬ |
the help of the newly developed NRMS technique. The four isomers a d can be produced from vinylacetylene, butatriene, 6,7-benzo-3-methylenetricyclo[3.2.2.024]nona-6,8-diene and 7,8-benzotricyclo[4.2.2.02,5]deca-3,7,9-triene, respectively49. Cyclobutadiene radical cation is also obtained directly from cis-3,4-dichlorocyclobutene557; its potential surface is calculated by Borden, Davidson and Feller558. McLafferty and collaborators554 show that neutrals that correspond formally to the condensation products of cyclobutadiene or methylenecyclopropene with benzene or naphthalene yield, on electron impact, C4H4Cž ions with a structure that corresponds to the C4H4 in the neutral. A summarizing overview is given by Zhang, Carpenter and McLafferty559.
The possibility of a DA reaction by the four isomers a d in Scheme 13 is studied in detail by Shay, Eberlin, Cooks and Wesdemiotis49. They use a pentaquadrupole instrument, where the Þrst (Q1), third (Q3) and Þfth (Q5) quadrupole are used as mass analyzers and the second (Q2) and fourth (Q4) as Ôreaction chambersÕ. Different combinations of the mass analyzing quadrupoles allow experiments that give answers to rather speciÞc questions such as sequential product and reaction intermediate scans. Although for none of the isomers is the DA-reaction product with isoprene detected at m/z 120, the authors are able to show that only cyclobutadiene d yields fragments that could be derived from the unstable intermediate product C9H12Cž . The authors formulate this ion as derived from a cycloaddition yielding the 2-methylcyclo[4.2.0]octa-2,5-diene (Scheme 14). The ions m/z 119 (loss of Hž ) and m/z 105 (loss of Mež ) form the basis of this conclusion. The latter ion can add an additional isoprene neutral in a second DA reaction to yield the ion m/z 173 (C13H17C ). In Scheme 14 a series of consecutive reaction products that are observed from these ÔprimaryÕ fragments of the DA product reinforce this conclusion. The origin of many of these ions is proven by using the multiple possibilities the pentaquadrupole instrument offers. The ion molecular reactions of the other ions a c with isoprene are dominated by charge exchange with the neutral, forming products starting from the isoprene cation (m/z 68), subsequently reacting with isoprene neutrals. The product spectra differ only slightly from the results of the ion/molecule reaction between isoprene ion with the isoprene neutral . These results show that the DA reaction has little chance in cases where competing reactions exist. The competition by the CT was conÞrmed when the isomeric ions were allowed to react with allene.
The correctness of these conclusions was proven by the fact that only cyclobutadiene underwent a DA reaction (series) with thiophene and furan49. Again the primary addition product is unstable, but the fragmentation products (loss of Hž and of Me) from the unstable precursor could again be identiÞed, in addition to the loss of C3H3ž , yielding the aromatic six-membered heterocyclic ring. In the reaction with the thiophene neutral the elimination of acetylene yielding an odd-electron ion is observed, necessitating a preceding rearrangement to a thiocyclohexadiene or thiocycloheptadiene structure before fragmenting. Other candidates as precursors (e.g. a substituted tiophenol) could be ruled out with the help of sequential product scans, showing again the possibilities of a multiquadrupole instrument. The authors49 conclude that it is difÞcult to distinguish the isomeric C4H4Cž ions on the basis of their ion/molecule reactions (a possibility offered by multisector instruments), whereas it is even more difÞcult to do so by conventional CID methods. This is both because of the milder conditions used to perform ion/molecule reactions and because bond-forming processes allow a greater variety of products than the (often) bruteforce conditions of CID. To determine the set of four C4H4Cž isomers one needs simply to apply the allene and thiophene reactions. The yields of C4H4Cž addition products to electronÐrich dienes decrease in the order d > a, b > c.
An important question is when the DA reaction is a concerted or a two-step cycloaddition. This question is answered experimentally by Groenewold and Gross560 and theoretically by Bauld and collaborators530,531 for the reaction of butadiene neutral with
2. Mass spectra of double-bonded groups |
67 |
C13 H17+
|
|
|
|
|
|
− Me• |
|
|
+ |
+ |
||||||
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
− C2 H2 |
||||||||
|
|
|
|
|
|
|
|
|||||||||
(C9 H12 )+ • |
|
|
|
|
|
|
|
C8 H9 |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
− H • |
|
|
|
|
|
|
|
|
− H2 |
|
|
|
|
− H2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
+ |
||||
C H + |
|
|
− CH4 |
|
C H + |
|
− C2 H2 |
|||||||||
9 |
11 |
|
|
|
|
|
|
|
|
8 |
7 |
|
|
|
|
|
|
− H2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
− C2 H2 |
|
|
|
|
|
|
|
|
|
|
|||
C H + |
|
|
C H |
+ |
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||
9 |
9 |
|
|
|
|
7 |
7 |
|
|
|
|
|
|
|||
− H2
C9 H7+
SCHEME 14
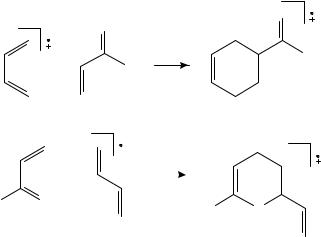
1,3-butadiene cation radical, and by Holman, Rozeboom, Gross and Warner561 with benzene ion in favor of the two-step process. In the former reaction the resulting C8H12Cž is decomposed by collision-induced fragmentation. If this ion is stabilized by collisions before CID, the resulting spectrum corresponds closely to the spectrum of vinylcyclohexene, but without collisional stabilization, a branched acyclic cation radical was found. Therefore the cyclization must proceed in two steps. CS2 is used as a stabilizing gas because of its high ionization energy and in order to avoid further reactions of neutral butadiene with the adduct. Slight deviations between the CID spectra of vinylcyclohexene and the collisionally stabilized product are explained by small amounts of [2 C 2] and/or [4 C 4] additions. The results with benzene are analogous: With collisional stabilization, the CID spectrum corresponds to a 2-phenyl-2-butene radical cation and without prior stabilization to 1-methylindan.
Chen and Williams562 demonstrated by ESR the rearrangement of 4-vinylcyclohexene to bicyclo[3.2.1]oct-2-ene in a CFCl3 matrix when irradiated by -rays and then
68 |
Tino Gaumann¬ |
allowed to warm from 77 K to 140 K. Semiempirical AM1 calculations by Dewar and coworkers563 have shown this rearrangement to be exothermic by 59 kJ mol 1. As an intermediate structure Chen and Williams propose either a bicyclo[3.2.1]oct-2-yl-6-ylium or a bicyclo[2.2.2]oct-2-yl5-ylium distonic ion. Vollmer, Rempel, Gross and Williams564 demonstrated the occurrence of this isomerization in the gas phase by use of MS/MS and ion cyclotron resonance spectrometry, using a specially developed technique to collisionally cool the ions565. When vinylcyclohexene and bicylo[3.2.1]oct-2-ene were separately ionized by electron impact, they had identical CID spectra. However, when they are ionized by charge transfer from CS2 and collisionally cooled, their CID fragmentation is different in the intensity ratio of the [MC Me]/[MC C3H6] peaks produced. Since the FT instrument allows a controlled excitation of the collisionally cooled ion, this peak ratio is measured as a function of the excitation energy. Since it passes through a maximum before decreasing to the small value corresponding to bicylo[3.2.1]oct-2-ene, the existence of an additional unstable and unknown intermediate ion is postulated.
1,3-Butadiene cation reacts with acrolein and methyl vinyl ketone to produce products that can be stabilized by collisions. The question arises as to whether the butadiene ion reacts as a dienophile or as an ÔeneÕ in these reactions? This problem has been attacked by Castle and Gross566. The two possible pathways are given in Scheme 15 (acrolein: R D H, a1, b1; Me vinyl ketone: R D Me, a2, b2). The authors use an ion cyclotron mass spectrometer at low pressures and a triple-sector instrument for CID studies at high pressures. Since they did not dispose of pulsed multiple inlet systems for their instruments, the simultaneous ionization of both components of the mixtures poses some problems. By measuring the time dependence of the product formation and disappearance and using the feature of double resonance in the FTICR instrument, they are able to circumvent some of the problems. An additional complication is the isomerization of the substituted pyran b1 to the aldehyde a1 by a Claisen rearrangement; the metastable decay of these two products are practically identical, as has been shown by Sarraf, Audier and Morizur567. Thus the two possible adducts a and b, that are not detectable at the low pressure of the FT instrument, are difÞcult to be characterized according to their CID spectra in the high
O
O
R
R
ÔdieneÕ |
ÔeneÕ |
|
|
|
|
(a) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R O
R O
ÔdieneÕ ÔeneÕ
(b)
SCHEME 15
2. Mass spectra of double-bonded groups |
69 |
pressure instrument. However, by a series of experiments the authors are able to advance arguments that the butadiene cation serves as the ÕeneÕ and the acrolein and methyl vinyl ketone react as dienes, forming adducts that have the pyran structure b. This is supported by the Þnding that a substituted pyran does undergo the corresponding RDA reaction in a metastable decay567.
At least in solution this does not seem to be the case, because Mayoral and collaborators568 performed the reactions between isoprene and acrolein and methacrolein in toluene solution and got only products that had the structure of a substituted cyclohexene carbaldehyde (a, b for acrolein; c, d for methacrolein in Scheme 16). Their aim was to study the RDA reaction of these products. The experimental results are complemented by semiempiric AM1 MO calculations, since it has been shown by Camaioni569 for the through-bond delocalization in bibenzylic systems and by Wiest, Steckhan and Grein570 for the calculations of the selectivity of the DA reactions in indoles and electron-rich dienes, that this method is perfectly useful for such systems. This is of importance for the decision, if under the assumption of a stepwise reaction the C(1) C(2) or the C(5) C(6) bond is opened, On the basis of these calculations, the authors could show that in the case of a the former and of b the latter case is energetically more favourable. This allows one to explain the observed differences in the electron impact and the metastable spectra of a and b: a ! [M Me]C , [M H2O]žC , [M CO]žC ; b ! [M CHO]C , as main peaks in the metastable decay. The case of c and d is slightly more complex. The calculations for this case show that the scission of C(1) C(2) is energetically more favorable than C(5) C(6) for both ions. This is consistent with the H2O loss for both isomers in the metastable fragmentation. By simplifying the results of the calculations one can say that a metastable loss of H2O is characteristic for a scission of the C(1) C(2) bond, whereas loss of ÐCHO is typical for an opening of the C(5) C(6) bond. Since [M HCO]C is much more important for d than for c, it is to be concluded that the opening of C(5) C(6) is more important for the former in spite of the calculations that predict a difference in heats of formation between C(1) C(2) and C(5) C(6) scission of 95 and 54 kJ mol 1 for c and d, respectively. Since the spectra are rather different, an isomerization of one form in another does not seem to be important, the problem remains to be deÞnitively resolved.
|
2 |
CHO |
CHO |
CHO |
CHO |
|
|
|
|||
3 |
1 |
|
|
|
|
4 |
5 |
6 |
|
|
|
|
|
|
|
|
|
|
(a) |
(b) |
|
(c) |
(d) |
SCHEME 16
The stereospeciÞcity of the RDA reaction can be used to produce unstable species and to determine their thermodynamic properties with the mass spectrometer. Such a project was undertaken by Turecekÿ and collaborators571 to distinguish the Z- and E-form of the dienol c given in Scheme 17. The 3-exo-vinylbicyclo[2.2.1]hept-5-en-2-ol is ßash-pyrolized in the inlet system of a mass spectrometer and ionized within a few ms after decomposition. The mass spectra and the ionization energies of E-c and Z-c were determined. The electron impact spectra are similar, although some reproducible differences can be seen; the CID spectra are identical. The same ions are produced by dissociative ionization of the precursors a and b. Their CID spectra are identical, however different from the isomeric aldehyde572. The ionization energies of the neutral dienols were measured as IE(E - c) D 8.51š 0.03 and IE(Z - c) D 8.47š 0.03 eV, respectively. Since the activation

70 |
|
Tino Gaumann¬ |
|
|
750 °C |
OH + |
|
|
10 −6 |
Torr |
|
OH |
|
|
(E)-C |
|
|
|
|
(a) |
|
|
|
|
|
|
OH |
|
750 °C |
+ |
|
OH |
10 −6 |
Torr |
|
(Z)-C
(b)
SCHEME 17
energy for thermal Z E isomerization has been estimated by Rabinovitch and Michel573
to be about 200 250 kJ mol 1, it is improbable that the two forms isomerize. The authors
obtain H°f [E - c]Cž D 733 š 5 kJ mol 1 and H°f [Z - c]Cž D 728 š 6 kJ mol 1. A possible isomerization before or during a RDA reaction must be taken into account.
With this question in mind Buchs and collaborators574 prepared a large number of D- and 13C-labeled 2-cyclohexen-1-ols and studied the loss of ethylene from the molecular ion which corresponds to the base peak in the EI spectrum. The mass shift for the different D-labeled compounds indicated no H/D scrambling, but the presence of (at least) two different RDA processes; 35% correspond to the elimination of C(5) C C(6) as neutral ethylene, in 65% C(4) C C(5) are lost. The latter route requires that the molecular ion undergoes an allylic isomerization either via a 1,3 shift of the C(1) hydrogen (shift of the double bond in position 1), or of the OH group. Similar isomerizations have been observed before, e.g. in cyclohexene297, menthene575 or methylcyclohexene576. The elimination of Me is not straightforward in 2-cyclohexen-1-ol. Labeling studies prove that the carbon of the Me group comes from position 2, whereas in the metastable time scale a complete scrambling of H/D is observed. Since Derrick, Falick and Burlingame297 have shown that on the ca 10 ps time scale of a Þeld ionization kinetics experiment, an All group of cyclohexene is eliminated without any scrambling, together with an additional H- atom, some sort of reversible isomerization between a sixand Þve-membered ring could be assumed. This has been shown to be the case for (even electron) c-Hex ions by Wesdemiotis, Wolfschutz¬ and Schwarz577. However, no deÞnitive conclusion could be reached for cyclohexenol.
Eberlin and Cooks578 discovered that acylium ions react with neutral isoprene and other 1,3-dienes in the gas phase to form covalently bound adducts by polar [4 C 2C ] Diels alder cycloadditions. The general reaction is given in Scheme 18, where R may range from H and alkyl to unsaturated, aromatic and polar substituents. The formation
O+ |
|
|
+ |
|
|
||
|
|
+ |
[4 + 2 |
+] |
O |
||
|
|
||||||
|
|
|
|
Me |
|||
|
|
|
|
|
|
||
|
|
|
|
|
|||
|
|
|
CID |
R |
|||
R |
|
|
|||||
SCHEME 18

2. Mass spectra of double-bonded groups |
71 |
of stable adducts occurs for the acetyl ion, but not for its C2H3OC isomers, as was also shown by the groups of Cooks579. Isoprene is used as neutral to check if a DA reaction (yielding a stable adduct or its fragments) or a charge or proton transfer takes place with the acyl ion. R D Me, Ph, Me2N, H2CDC(i-Pr), PhHCDN yielded stable adducts in at least tenfold excess compared to the proton-transfer product; for R D H, MeNH, HO, Cl only the CT product could be observed. Some substituents show comparable probabilities for both reactions. A second question is the necessary structure of the neutral indispensable for a stable adduct. The acetyl ion is used as probe. Only isoprene, 1,3-butadiene and cyclopentadiene give a stable adduct whose structure seems to correspond to a [4 C 2C ] addition. When 3-methylenecyclohexadiene is used, with a diene which is locked in the trans conÞguration, the adduct is not generated and the product spectrum is dominated by proton transfer. It seems that a cis-diene conformation is needed for the DA reaction. In order to discriminate against a [2 C 2C ] addition, the reaction of 1,3-butadiene is compared with that of limonene, a nonconjugated diene. The latter yields very little adduct products. Similarly, the acetyl cation fails to show substantial adduct formation with benzene, benzonitrile and Et vinyl ether. Thus, ample evidence is produced to show that a [4 C 2C ] addition takes place. MS3 spectra (see above) give additional evidence for such an addition.
The reaction of vinyl Me ether cation radical with 1,3-butadiene forms another case where the reaction scheme seems to be rather complicated, as is demonstrated by Groenewold and Gross580. When the reaction is being conducted at low pressure, no adduct is observed. Since the ether as well as the diene is ionized by electron impact, both molecular ions and their fragmentation products are observed in the ion source. The time revolution in the ICR spectrometer shows a rapid transfer from the butadiene ion to vinyl Me ether. At the same time C6H8Cž (m/z 80) and C6H9OC (m/z 97) rise in a ratio of about 10:1. The fragment m/z 80 corresponds to a loss of methanol, typical for a 4-methoxycyclohexene, as is shown by deuterium labeling by van Doorn, Nibbering, Ferrer-Correia and Jennings581. These observations are strong evidence for a cyclic adduct at low pressures. In a high-pressure ion source the adduct ion is stabilized by collisions; it can be isolated and studied by CID. There is no resemblance with any cyclic product, but a nearly 1:1 correspondence with the spectrum of 2,4-pentadienyl Me ether. Therefore the authors propose the reaction sequence of Scheme 19, where an acyclic unstable
|
O |
|
* |
|
|
|
O |
+ |
acyclic * |
||
C |
H |
+ |
|
|
7 |
12 |
|
|
|
N |
− CH3 OH |
|
|
|
|
|
|
|
O |
SCHEME 19

72 |
Tino Gaumann¬ |
intermediate adduct either undergoes ring closure to the unstable substituted cyclohexene or is stabilized by collisions to the stable pentadienyl Me ether. Sequential and not two parallel reactions are preferred, because in the latter case, as well as in the lowas in the high-pressure spectra, evidence of both structures should be evident, which is deÞnitely not the case.
The inßuence of stereochemistry on the mass spectrum in the RDA reaction can be well studied with substituted norbornanes (bicylo[2.2.1]heptane). Traldi, Cativiela and collaborators582 studied the case of monoand disubstituted norbornanes (see Scheme 20). For the carboxylic acids 1a and 2a, there is a clear difference in the spectra produced by electron impact: e.g. the endo-form 1a having [M C2H3O2R]Cž as the base peak, whereas the exo-form 2a had a much stronger tendency to lose the carboxylic group. However, this is not the case for the slow metastable decay, where spectra of both ions are indistinguishable. The isomerization of the two forms, although slow compared to the larger part of the decay (which takes place in <1 ns), is faster than the metastable time window (>1 s). Clear differences are also observed for the two isomers 1b and 2b, but in contrast to 1a and 2a, also the metastable spectra are different, e.g. the intensity of [M ROH]Cž is more important for the exo-isomer. 1c and 2c are again different in the electron impact and the metastable spectra, but for 1d and 2d only minor differences can be seen in both kinds of spectra. Different substituents may change the fragmentation pathways and with this a stereoisomeric difference may manifest itself or not. Of special interest is the dependence on the time scale of fragmentation; isomerization may precede the relatively long time needed for a collisional excitation experiment. However, the fact that the RDA reactions of differently D-labeled 5-norbornenols have been used by Turecekÿ and Hanusÿ583,584 to prepare by ßash pyrolysis differently labeled unstable ethenols and to obtain their mass spectra proves that usually the fast fragmentations proceed without too much isomerization.
|
|
O |
|
|
|
R |
O |
|
|
|
|
|
||
O |
O |
R |
|
|
R |
′ |
|||
|
|
|||
(1) |
R′ |
(2) |
|
|
|
|
(a)R= R′ = H
(b)R= H;R′ = CH3
(c)R= NHCOCH3 ;R′ = H
(d)R= NHCOCH3 ;R′ = CH3
SCHEME 20
Vainiotalo and collaborators585 measured the electron impact spectra of several either di- exo- or di-endo-norbornane/ene-fused 2-NÐphenyl iminoperhydro-1,3-oxazines. Whereas the stereoisomeric unsaturated compounds cannot be distinguished on the basis of their electron impact spectra, the spectra of the stereoisomeric saturated compounds are sufÞciently different to allow differentiation of the stereoisomers. The N-substituents were either H or Me. The difference is due to the fact that the unsaturated compounds fragment mainly by two RDA reactions with Þrst a loss of cyclopentadiene and then yielding