

Supplement A3: The Chemistry of Double-Bonded Functional Groups. Edited by Saul Patai Copyright 1997 John Wiley & Sons, Ltd.
ISBN: 0-471-95956-1
CHAPTER 6
Dipole moments of compounds containing double bonds
OTTO EXNER
Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Prague, Czech Republic
Fax: +422-243-100-90; e-mail: uochb@uochb.cas.cz
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
261 |
II. PROGRESS IN THE DIPOLE MOMENT METHOD . . . . . . . . . . . . . |
264 |
A. Experimental Procedures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
264 |
B. Calculations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
265 |
III. CDC BONDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
266 |
IV. CDN BONDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
269 |
A. Isolated CDN Bonds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
269 |
B. Conjugated Systems X CDN . . . . . . . . . . . . . . . . . . . . . . . . . . . |
270 |
C. Conjugated Systems CDN X . . . . . . . . . . . . . . . . . . . . . . . . . . . |
272 |
D. Conjugated Systems X CDN Y . . . . . . . . . . . . . . . . . . . . . . . . |
274 |
V. CDO BONDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
280 |
A. Isolated CDO Bonds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
280 |
B. Conjugated and Cumulated Systems C CDO . . . . . . . . . . . . . . . . |
281 |
C. Conjugated Systems O CDO and S CDO . . . . . . . . . . . . . . . . . . |
285 |
D. Conjugated Systems N CDO . . . . . . . . . . . . . . . . . . . . . . . . . . . |
288 |
E. Conjugated Systems X CDO . . . . . . . . . . . . . . . . . . . . . . . . . . . |
293 |
VI. CDS BONDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
294 |
VII. CDP BONDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
298 |
VIII. OTHER DOUBLE BONDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
298 |
IX. REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
301 |
I. INTRODUCTION
Compounds with double bonds are important for the theory of dipole moments and dipole moments are of importance for determining structure of compounds with double bonds. There are two reasons for this. All double bonds give some degree of rigidity to the molecule which enables the partial dipole moments to be treated as vectors of known
261

262 |
Otto Exner |
direction. Second, some double bonds, particularly CDO and CDN, themselves possess a considerable dipole moment of known direction and of a reasonably constant magnitude. Therefore, dipole moments were one of the first physical methods applied for determining configuration on the double bond or conformation on the adjacent single bonds (or partial double bonds). The example of (E)- and (Z)-1,2-dichloroethenes1 may be considered as the first in history when configuration was determined directly and nonempirically, from a pure principle. Numerous further examples were reviewed in a previous article2 in this series. Particularly important was the extension to partial double bonds whose double-bond character is sufficiently strong to keep the molecule in a relatively stable conformation. In this case dipole moments are often an efficient experimental approach (esters, amides).
Nevertheless, when this article is continued after two decades, it should not remain restricted to stereochemical problems. During that time, other very powerful methods will have been developed which are mostly easier and often also more reliable. Particularly, the recent development in X-ray analysis gives a completely reliable proof of configuration valid even for the isolated molecule: in the case of conformation it may appear necessary to prove that it is unchanged in solution (see for instance Reference 3). While X-ray is the most reliable method, NMR spectroscopy is the fastest. It still uses some empirical rules and comparison with model compounds, but in a modern version (2-D NMR, NOE) it is also completely trustworthy. Therefore, many recent dipole-moment studies investigated compounds whose steric arrangement was already known, and attention was focused on the electron distribution on the individual bonds, or in conjugated systems. The difference in the point of view may be explained as follows.
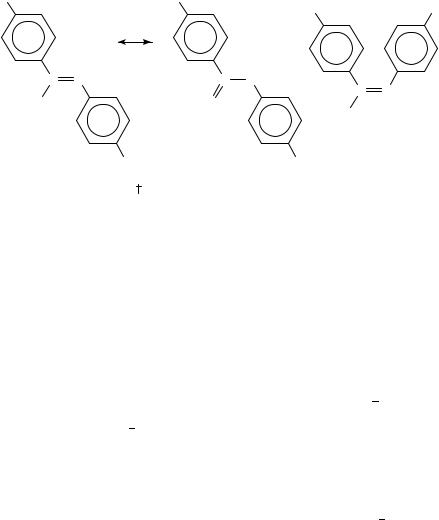
The dipole moment of a molecule can be generally interpreted in one of two ways. If we know with a certain approximation the electron distribution on the individual parts of the molecule, expressed say in terms of partial dipole moments or bond moments, then we can determine the mutual position of these parts to each other. The other possibility is that if we know the steric arrangement, i.e. configuration and conformation, then we can determine the electron distribution in some of the parts and express it, for instance, in terms of resonance formulas, of special moments valid only in this molecule, etc. Thus, in 1933 the dipole moments of substituted azoxybenzenes (1, 2) were first used for determining their configuration. Although the reasoning was not exact with respect to modern views, the result was right4. The simplest proof today would be a reference to the 4,40 -dichloro derivative. Since 1a and 2a possess similar dipole moments, quite different from 1c, the configuration Z is assigned to 1a.
Fifty years later, when the configuration was known beyond any doubt, the dipole moments were reinterpreted in terms of electron distribution5. The direction of was determined for 1a by means of substituted derivatives; the same was done for (E)- azobenzene as reference. The vector difference between m of these two compounds should
represent the bond moment N O. The standard NC O bond moment† was derived from trimethylamine oxide and the dipole moment anticipated for the ideal structure 1a was calculated; the vector difference against mexp was called the mesomeric dipole moment mm according to the vector equation 1. This finding was explained by mesomerism of
the two formulas 1a and 1b. Since the derived mm was not oriented† from O toward N(2) but merely from O toward N(1), it was concluded that these mesomeric formulas do not describe the electron distribution completely and a hypothesis was advanced that the nitrogen atom N(1) could allocate more than its equivalent of eight electrons5. This
† Direction of the dipole moment is shown always from the positive toward the negative end; in the bond moments the first atom is positive. Units D (debye) are used throughout for better comparison with the previous review2; 1 D D 3.334 10 30 C m.
|
6. Dipole moments of compounds containing double bonds |
263 |
X |
X |
|
|
X |
X |
|
N+ N |
N+ N− |
N+ N |
|
O− |
O |
|||
O− |
||||
|
|
|
||
|
X |
|
X |
|
(1a) |
X = H exp 1.70 |
(1b) |
(1c) exp 4.71 |
|
(2a) |
X = Cl exp 0.96 |
|
|
|
hypothesis need not be accepted: the example should only show how the dipole moments can be exploited in two different ways.
mm D mexp mcalc |
1 |
In the examples which will follow, usually only one of the two problems is dealt with, although we do not exclude solving both problems only from the dipole moment values. Due to the competition of other methods, the stereochemical applications are suitable mainly for compounds with many heteroatoms and unstable conformations (unsuitable for NMR). Estimation of conjugation remains specific for dipole moments but is only a formal approach and may become, in some cases, too abstract. Actually, the two interpretations appear together rather often, and for this reason they will not be separated in this article either. Instead, the material will be divided according to individual bonds: n conjugated systems will be arranged according to the main, formally double bond (for instance amides under the CDO bond), conjugated systems according to the bond which is dealt with later. The whole material covered exceeds a little that delimited for this Volume but corresponds to that in the previous article2.
Let us mention that there are still other relations between conformation and dipole moments. Relative stability of conformational isomers was often brought in connection with their dipole moments. For instance, the preferred sp conformation of esters (3a), or of carboxylic acids, was often explained by the lower dipole moment6 compared to the conformation 3b, or in other words by the electrostatic repulsion7 9 of partial dipoles in 3b. Reasoning of this kind is usually based on anticipated rather than measured moments: that of 3b cannot be directly measured. Interaction of more distant dipoles is commonly neglected, viz both its effect on the conformation and on the total (see, for example, Klinot and coworkers10). This is in fact the fundamental assumption underlying all analyses of dipole moments.
Dipole moments of solutes are also involved in the so-called reaction-field theory11,12 which predicts generally the Gibbs energy of solvation, and from it the stability of conformers as dependent on solvents. Besides the dipole the quadrupole moment is also taken into the calculations. For instance, conformational equilibria of cyclic halo ketones were predicted from the dipole moments of the two conformers with fair success13. However, the whole theory was criticised14,15 that there is too much arbitrariness in the
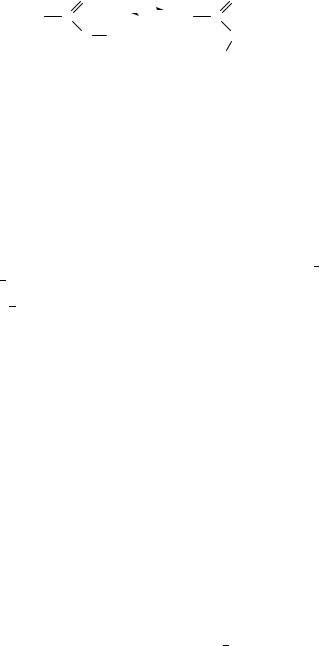
264 |
Otto Exner |
|
||
O |
|
|
|
O |
R1 C |
|
|
R1 |
C |
|
|
|||
|
|
|||
O |
R2 |
O |
||
|
|
|
|
R2 |
m calc 1.6 |
|
|
|
m calc 3.4 |
(3a) |
|
|
|
(3b) |
parametrization, so that almost any experimental result can be fitted. In any case, it is true in a qualitative sense that the form with a larger dipole moment is preferred in a polar solvent (see, for instance, Malecki and Nowak16).
II.PROGRESS IN THE DIPOLE MOMENT METHOD
A.Experimental Procedures
Concerning the dipole moments in general theory, experimental determination and applications in chemistry, see References 17 and 18. Since our last review2 there has been little progress in this respect. The common experimental methods were compared and almost no difference was found between the two mostly used (Halverstadt Kumler and Guggenheim Smith)19. New methods of calculating from standard permittivity measurements were advanced20, which particularly allow extending the measurements to polar solvents21 23. They have not been used more broadly: all data cited in this review were obtained in nonpolar solvents or in the gas phase. On the other hand, in nonpolar solvents problems may arise with dimerization of certain compounds: the calculations used until now were corrected from the statistical point of view24. These problems may be avoided by working at sufficiently low concentrations, although for some compounds (such as carboxylic acids) reliable values for the monomer cannot be obtained. The correction term for atomic polarization was also reinvestigated. Although it is still at issue25, particularly in the case of certain symmetrical compounds and their apparent small dipole moments26,27, the standard correction of 5% is recommended as a mean value for compounds of various structures19.
A simplified experimental method which avoids measuring both density and refractive index (the Higasi method) has attracted new attention and was recommended28, modified29 and a new similar procedure advanced30. More trustworthy in our opinion is a statistical test31 which concluded that the Higasi method is both inaccurate and biased and can be of acceptable accuracy only for compounds with rather high dipole moments.
Dipole moments determined in the gas phase by microwave spectroscopy are always accepted as reliable standards. However, even in this method errors may arise32. Moreover, this approach has remained restricted to rather small molecules. For the simplest molecules the method of molecular beam electric resonance can yield actual standards33, by three orders more accurate than common methods. A program was started for measuring standard values of in solution at conditions similar to the gas phase34. A high-frequency technique allowed working in a nonpolar solvent (cyclohexane) at high dilution (weight fraction 10 4). However, only few data are available at present and their accuracy is not particularly high.
We may conclude that the dipole moments determined 20 30 years ago by standard methods either in solution or in the gas phase are directly comparable to those measured

6. Dipole moments of compounds containing double bonds |
265 |
today in the same state. Older data in solution must usually be recalculated; see, for instance, Reference 5.
B. Calculations
Dipole moment for a given structure can be predicted by at least four methods: ab initio, semiempirical calculations, molecular mechanics and the bond moment scheme. At the present state of development, one cannot say generally which is preferable. The ab initio methods require a rather large basis and/or special sophisticated methods35 37, so that they are of limited importance for molecules of that size which are dealt with in this review. Even so, the results are of different reliability in individual cases38 40. As an exception, even a simpler variant with a small basis may work41. One of the most difficult problems for calculations seems to be the too large moment of the nitro group41 which is so popular in experimental work. Recently, the 6-31G(CsdCsp) basis was recommended42 specially for dipole moments but the examples given represent molecules that are still small for our purposes. The ab initio methods can be combined with the reaction-field theory to calculate in different solvents43,44; experimental verification is difficult.
It is thus not surprising that semiempirical methods are still in use, either as standard methods45 49 or in numerous special modifications50 57 advanced for dipole moments, sometimes still for a particular class of compounds such as the second-row elements55, nitrogen56 or phosphorus57 compounds. All these variants may possibly work, each in a certain range of derivatives. Sometimes, the results are quite satisfactory47, in other cases quite bad48,49,51, for instance particularly for benzene derivatives with polar substituents49 which are so important in the common analysis. Various semiempirical methods were compared many times45,46,50 53 but it is not possible to say which is generally preferable.
Molecular mechanics has often been found satisfactory within a limited set of compounds. One possible approach, called the IDME method13, can be viewed as addition of bond moments improved by an additional inductive term. By incorporating the concept of reaction-field theory, effective in various solvents can be obtained. The examples given concern mainly halogen and oxo derivatives, particularly of cyclic structures13. The second approach, elaborated more extensively, is the proper molecular mechanics in the MM3 force field58. Within its framework can also be obtained as a merely marginal result: calculation is also based on bond moments but uses more differentiated values (compared to the simple vector addition mentioned below). Some parameters must be determined for every class of compounds separately. Of the compounds dealt with in this review, particularly aldehydes and ketones58, diketones59, carboxylic acids and esters60, amides61 and oximes62 were elaborated. Each extension needs a couple of new empirical parameters and, with a small number of compounds, it is difficult to assess the success of this method. In a somewhat strange approach63 the geometry is obtained from MM, then
calculated ab initio.
Last but not least, simple vector addition of bond moments still remains a method of
choice in most structural applications. In detailed studies on rigid model molecules it was found that its principle is not quite exact64 but the deviations from additivity were of a size which can be neglected in routine work. Particularly, a constant C H bond moment was criticised as an oversimplification65,66 since of hydrocarbons is not zero67. Nevertheless, the system works reasonably in practice and may still be improved when the bond or group moments are determined and applied in a certain range of similar compounds. A refinement taking into account the mutual polarization of all bonds was already mentioned13. This works for halogen and oxo derivatives, but it is not applicable for most compounds dealt

266 |
Otto Exner |
with here. Its simplified version, in the form of a computer program, should serve merely for demonstrative purposes68. Nevertheless, a deviation from the additive behavior may be appreciable even in monofunctional derivatives when a strongly polar group is bonded to a larger hydrocarbon residue69. Therefore, in detailed studies it is better to compare only molecules of similar size. In our opinion, the addition of bond moments is the method of choice, particularly when the effect of a remote substituent is to be accounted for. It works particularly well for substituents on the benzene ring which are used as references for determining the direction of other moments in the same molecule. This approach has been proved in numerous examples2. Sometimes, the concept of bond moments need not be used explicitly and the anticipated values of can be taken directly from model compounds10.
For determining conformation from the experimental dipole moments, two methods in common use may be considered as standard17. In the first, dipole moments of several para-substituted aromatic derivatives are measured70. In the second, the dipole moment is combined71 with the Kerr constant, mK; one can make do with a single compound, but evaluation of the Kerr constant may be too complex and sometimes not completely reliable. In either case, a graphical representation is useful: either 2 values are plotted of substituted vs unsubstituted derivatives70, or 2 vs mK71. In the first method, it has been commonly assumed that a distant substituent contributes an additional component to the overall dipole moment but does not itself influence the conformation. Many examples
were given in the previous review2 mostly an acceptable approximation,
and further ones are given here. This principle is but it has also its limitations as revealed in one
example by comparison with more accurate NMR results72.
III. C=C BONDS
Unsymmetrical olefinic hydrocarbons possess very small dipole moments which nevertheless can be measured in the gas phase by microwave spectroscopy67. Within the framework of bond moments17, these values are usually accounted for by attributing different values to the C H bonds according to the hybridization on carbon: H Csp2 may be taken con-
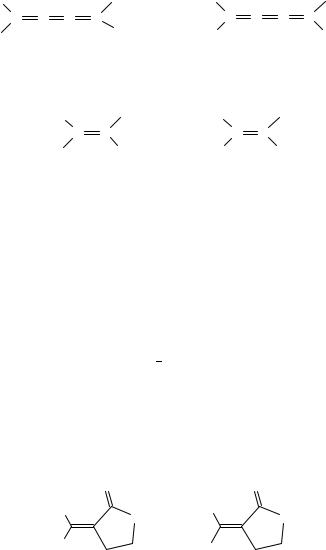
ventionally as zero, then H Csp3 equals 0.3 D. However, the dipole moment of styrene73 (0.12 D) cannot be included in this scheme and should be interpreted either by a nonzero value for the Car Csp2 bond, or by different values for H Csp2 and H Car. In common calculations from bond moments, such details are neglected. With the conjugated systems 4 6, was compared to similar nonconjugated hydrocarbons74. The result was that 4 has an aromatic character, 5 and 6 do not. Compound 7 has a very high 75; the aromatic character of the two rings is obvious.
mexp |
1.90 |
m exp |
0.42 |
mexp 0.48 |
m exp 3.71 |
mcalc |
2.34 |
mcalc |
0.43 |
mcalc 0.49 |
|
(4) |
(5) |
(6) |
(7) |
||
6. Dipole moments of compounds containing double bonds |
267 |
||||
H |
|
t-Bu |
t-Bu |
|
t-Bu |
C C |
C |
C |
C |
C C C |
|
t-Bu |
|
Cl |
H |
|
Cl |
|
|
|
|||
mexp |
1.7 |
|
|
mexp 2.3 |
|
(8a) |
|
|
(8b) |
|
|
H |
|
t-Bu |
t-Bu |
t-Bu |
|
|
C |
C |
C |
C |
|
t-Bu |
|
Cl |
H |
Cl |
|
|
mexp 2.0 |
mexp 2.3 |
|
||
|
|
(9a) |
|
(9b) |
|
For determining configuration on a CDC bond, the dipole moment approach is not of much use. An exceptional case was reported76 concerning the configuration on the cumulated double bonds in the 1,2,3-butatriene derivatives 8a,b. The long-range coupling constants, 5JHH, have not brought any decision and other methods were prevented by the properties of these compounds (rather unstable liquids). Resolution made on the basis of dipole moments exceeds the framework of bond moments: within it the dipole moments of 8a and 8b should be equal. In the second approximation, one can consider also the polarization of the hydrocarbon chain by the C Cl dipole: then greater is anticipated for 8b. This assumption was checked on simple ethene derivatives 9 used as model compounds, then the configuration of 8a and 8b was deduced from experimental dipole moments.
When a CDC bond is involved in an n conjugation, the electron distribution must be taken into consideration together with the conformation. In the compound 10 the CDC bond is conjugated with an ester group but the problem was configuration on CDC. It was assigned on the basis of dipole moments77; their anticipated values were obtained from bond moments with variable conformation on C O. The only possible solution was finally ap conformation on C O and zero m. Agreement with experiment is not good: assignment was made merely in a qualitative sense by comparing the two isomers. In this case, there were evidently possibilities of assignment also by other methods.
O |
|
|
O |
CH3 |
O |
RO |
O |
|
|
||
RO |
|
CH3 |
|
mcalc 3.0 |
|
mcalc |
5.4 |
mexp 1.71 |
|
mexp |
4.86 |
(10a) |
|
(10b) |
|
Dipole moments of nitrovinylamines 11 were larger than the sum of bond moments and the simple algebraic difference was taken as a measure of through-conjugation78. Conformation was assumed as planar. In this way, the exact direction of the mesomeric momentm remained unknown: evidently the structure of the compound would not allow one
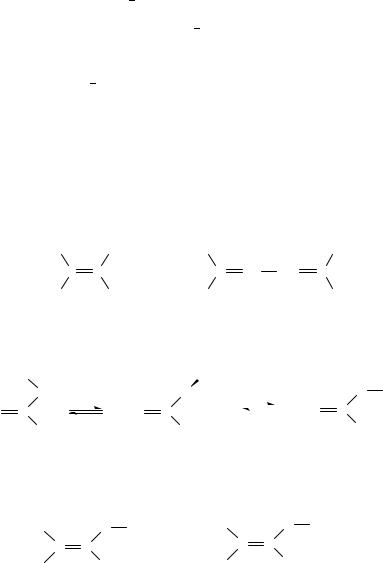
268 |
Otto Exner |
to introduce the necessary substituents. When the conjugated chain is extended (12), the calculated dipole moment increases79 with the number of vinyl groups up to n D 24: the calculated values are overestimated but the trend could be reproduced well. As expected, the high dipole moments are strongly reduced, when the chain is rotated by 90° around one single bond80. Further push pull systems were studied theoretically81, some of them containing also benzene rings instead of ethene units79,82,83. In the case of vinyl ethers and vinyl sulfides, neither the conformation nor n conjugation can be neglected. According to IR spectra and from the temperature dependence of , an equilibrium of the sp and sc (<CDC O C 90°) forms (13a,b) was assumed for methyl vinyl ether84, sc is populated at higher temperatures and prevails in t-butyl vinyl ether. The mesomeric moment of 0.72 D expressing the n conjugation was obtained previously from model compounds (for methyl vinyl sulfide 0.24 D). However, this result is at variance with calculations85 claiming equilibrium of the sp and nearly planar ap conformations (13a,c). A more reliable analysis was carried out in the case of the dichloro derivatives 14 and 15. It started from the assumption that the t-butyl ether 15 must be in the ap conformation86: no minor form was detected by IR spectroscopy. The dipole moment of 15 was measured and its direction estimated by means of the experimental Kerr constant. From it a group moment of the Cl2CDCH residue was evaluated and from it the dipole moments anticipated for the methyl ether 14 in various conformations; the same was done for the Kerr constant. The result was that even the methyl ether 14 (and ethyl and i-propyl ethers as well) is in the ap conformation. This example shows a very ingenious combination of various methods.
H |
NO2 |
H |
|
|
|
NO2 |
C |
C |
C( |
CH |
|
CH)n |
C |
R1R2 N |
H |
(CH3 )2 N |
|
|
|
H |
µ calc |
2.3 |
µ calc |
11.94−25.52 |
|
||
(11) |
|
|
(12) |
|
|
|
H3 C |
|
CH3 |
|
|
|
|
O |
|
O |
|
|
|
O CH3 |
CH2 C |
CH2 |
C |
|
|
CH2 C |
|
|
|
|||||
|
|
|||||
H |
|
H |
|
|
|
H |
m calc 1.11 |
|
mcalc 1.88 |
|
|
|
|
mexp 1.11 |
|
|
|
|
|
|
(13a) |
|
(13b) |
|
|
|
(13c) |
Cl |
O CH3 |
Cl |
|
|
O |
Bu-t |
C |
C |
C |
C |
|
||
Cl |
H |
Cl |
|
|
H |
|
mcalc |
2.77 |
|
mexp |
3.18 |
|
|
mexp 2.74 |
|
|
|
|
|
|
(14) |
|
(15) |
|
|
||
6. Dipole moments of compounds containing double bonds |
269 |
The Cl2CDCH group moment was not further resolved and no mesomeric moment was calculated. Ethenethiol has sp conformation in the gas phase as determined by MW and IR spectroscopies; at this opportunity the dipole moment was also measured87.
With 3-methoxypropenenitriles (16, 17) the problem was still more difficult since the conformation and conjugation were both estimated from dipole moments only88. Similarly, as in the preceding example, the Z isomer was taken as a fixed model which can have only ap conformation. Then the difference between the calculated and experimental moments was attributed to n conjugation. In the case of the E isomer, the conformation sp seems possibly somewhat preferred but, irrespective of the conformation, the conjugation is strong, stronger than in the isomer Z. Again the analysis is incomplete since the directions of the mesomeric moments are not known exactly.
|
|
H3 C |
|
|
|
H3 C |
||
|
|
|
|
|
|
|
||
NC |
O |
NC |
|
O |
H |
O |
H |
O |
|
|
CH3 |
|
|
|
|
CH3 |
|
C |
C |
C |
C |
|
C |
C |
C |
C |
H |
H |
H |
|
H NC |
H |
NC |
H |
|
mcalc |
4.50 |
mcalc |
3.06 |
mcalc 3.12 |
mcalc 3.24 |
|||
mexp 4.98 |
|
|
|
|
|
mcalc 4.38 |
|
|
(16a) |
(16b) |
|
(17a) |
(17b) |
||||
In the conjugated system CDC N the configuration on N can be assumed as planar. Then the conformation can be unambiguous and attention is focused on the electron redistribution due to conjugation. For instance, in 18 the conformation was not exactly known; conjugation was expressed89 by the mesomeric moment of 0.9 1.5 D, calculated in a rather complex way.
N
(18)
An interesting example of exploiting the dipole moment of simple olefins is the equilibrium between (E)- and (Z)-1,2-dichloroethenes in various solvents90. Equilibrium constants were determined indirectly (without achieving equilibrium conditions) and correlated with the solute dipoles and quadrupoles within the framework of reaction-field theory11 with relative success.
IV. C=N BONDS
A. Isolated C=N Bonds
The CDN bond appears as most interesting from the point of view of dipole moments: it has itself a considerable moment, gives rise to variable configurations and can be involved in many conjugated systems. Nevertheless, an isolated CDN has been dealt with relatively rarely. Configurational equilibrium of some azomethines was investigated91 mainly with the idea to compare the possibilities of dipole moments and of NMR spectroscopy. In the 4-chloro derivatives 19, the substituent enabled the equilibrium to be determined
270 |
Otto Exner |
independently and nonempirically. However, NMR spectroscopy gives this equilibrium more precisely. With some other azomethines, the dipole moment of the equilibrium mixture was calculated using the NMR results and was in agreement with experiment91. In the azomethines 20 the dipole moments are more telling for the configuration, due to the presence of the polar CN group92: only the E configuration is present.
R |
CH3 |
R |
NC |
C |
N |
C N |
C N |
|
|
CH3 |
Ar |
X |
X |
X |
X = H, Cl |
|
|
R = CH3, C2H5, i-C3H7, t-C4H9 |
|
|
(19a) |
(19b) |
(20) |
In the more complex azomethines 21 the configuration on CDN was in fact known from NMR; dipole moments served mainly to determine the ap conformation on the Car C(N) bond93. With the simple fluorinated azomethine 22 there are no stereochemical problems. The detailed geometry was determined94 by both electron diffraction (ED) and by microwave (MW), and the dipole moment was also measured but not interpreted; even its direction is not known with certainty.
H |
|
CH(CH3 )C6 H5 |
|
C |
N |
F |
F |
N |
|
||
|
C |
N |
|
|
|
||
|
|
F |
|
|
|
mexp 1.41 |
|
(21) |
|
|
(22) |
B. Conjugated Systems X−C=N
Structural chemistry of amidines is complex, including problems of tautomerism, configuration on the CDN bond, conformation on the C N bond and electron distribution in the conjugated system. These problems were often mixed together in the literature but may be solved separately on suitable model compounds. In the case of N,N,N0- trimethylbenzamidines (23) it is in principle possible to deduce both the configuration and extent of resonance (23a $ 23b) solely from dipole moments95. These possibilities are shown in Figure 1, which may serve also generally as an example of this kind of dipole moment analysis. The direction of the experimental dipole moment was determined by triangulation from the moments of the unsubstituted amidine (HX), its 4-nitro derivative (NX) and the nitro group itself (NH). Of the vectors calculated for the configurations (E and Z), none matches the experimental vector exactly: the difference would represent the mesomeric dipole moment mm according to equation 1. Now, the