

66. Ректификация. Области применения, аппаратурное оформление и основные отличия от простой перегонки.
РЕКТИФИКАЦИЯ (от позднелат. rectificatio - выпрямление, исправление), разделение жидких смесей на практически чистые компоненты, отличающиеся т-рами кипения, путем многократных испарения жидкости и конденсации паров. В этом осн. отличие ректификации от дистилляции, при к-рой в результате однократного цикла частичное испарение -конденсация достигается лишь предварительное (грубое) разделение жидких смесей.
Для ректификации обычно используют колонные аппараты (см., напр., Насадочные аппараты, Тарельчатые аппараты), наз. ректификационными колоннами, в к-рых осуществляется многократный контакт между потоками паровой и жидкой фаз. Движущая сила ректификации-разность между фактическими (рабочими) и равновесными концентрациями компонентов в паровой фазе, отвечающими данному составу жидкой фазы. Парожидкостная система стремится к достижению равновесного состояния, в результате чего пар при контакте с жидкостью обогащается легколетучими (низко-кипящими) компонентами (ЛЛК), а жидкость - труднолетучими (высококипящими) компонентами (ТЛК). Поскольку жидкость и пар движутся, как правило, противотоком (пар-вверх, жидкость - вниз), при достаточно большой, высоте колонны в ее верх. части можно получить практически чистый целевой компонент.
В зависимости от т-р кипения разделяемых жидкостей ректификацию проводят под разл. давлением: атмосферным (т. кип. 30-150 °С), выше атмосферного (при разделении жидкостей с низкими т-рами кипения, напр. сжиженных газов), в вакууме (при разделении высококипящих жидкостей для снижения их т-р кипения). Ректификацию можно осуществлять непрерывно или периодически. Для непрерывной ректификации применяют колонны, состоящие из двух ступеней: верхней-укрепляющей (в ней пар укрепляется, т.е. обогащается ЛЛК) и нижней - исчерпывающей (где происходит исчерпывание жидкой смеси, т. е. извлечение ЛЛК и обогащение ее ТЛК). При периодической ректификации в колонне производится только укрепление пара. Различают ректификацию бинарных (двухкомпонентных) и многокомпонентных смесей.
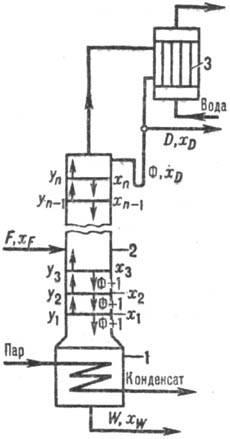
Рис. 1. Ректификационная установка непрерывного действия: 1 -куб-испаритель; 2-колонна; 3-дефлегматор.
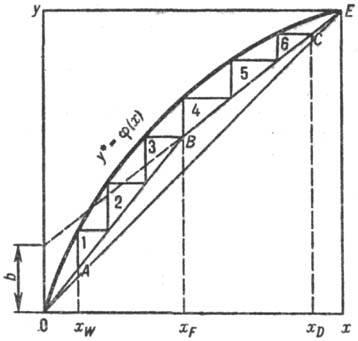
Рис. 2. Графическое определение числа теоре-тич. тарелок; ОE-равновесная кривая; АВ и ВС- рабочие линия для укрепляющей в исчерпывающей частей колонны; 1-6-тарелки.
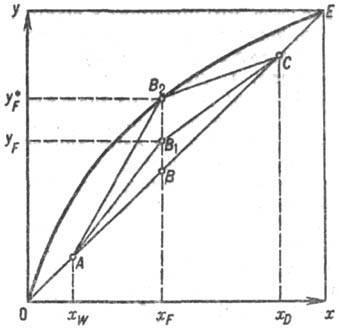
Рис. 3. Положение рабочих линий непрерывной ректификации на у-х-диаграмме.
Ректификация бинарных смесей Процесс осуществляют при дискретном (ступенчатом) контакте фаз в тарельчатых колоннах или непрерывном контакте фаз в насадочных колоннах.
Непрерывная ректификация. При ректификации в тарельчатых аппаратах (рис. 1) исходная смесь в кол-ве F с концентрацией низкокипящего компонента xF поступает (здесь и далее в единицу времени) в среднюю часть колонны; верх. продукт-дистиллят в кол-ве D с концентрацией низкокипящего компонента XD отбирается из дефлегматора, а обедненный этим компонентом остаток в кол-ве W с концентрацией xw отводится в качестве ниж. продукта из куба-испарителя (F, D, W-в моль/ч, Хр, XD, XW-B молярчых долях). Образующиеся в нем пары поднимаются по колонне, контактируя на тарелках от 1 до п со стекающей жидкостью, и поступают в дефлегматор, откуда часть образовавшегося конденсата, наз. флегмой (Ф моль), возвращается в верх. часть колонны.
Материальный баланс по ЛЛК для всей колонны имеет вид:
![]()
При работе колонны в адиабатич. условиях и равенстве молярных теплот испарения компонентов в каждом сечении укрепляющей части (выше ввода питания) концентрация ЛЛК в паре связана с его концентрацией в жидкости х ур-нием т. наз. рабочей линии:
![]()
где R = Ф/D - флегмовое число. Ур-ние рабочей линии для исчерпывающей части колонны (ниже ввода питания):
![]()
где f = F/D. Зависимость между предельными, или равновесными, концентрациями распределяемого в-ва в фазах изображается графически т. наз. равновесной линией.
Для анализа работы колонны, расчета состава дистиллята и остатка и распределения концентраций ЛЛК по высоте аппарата используют понятие о теоретической ступени разделения, или теоретической тарелке (ТТ). Такая ступень (тарелка) соответствует нек-рому гипотетич. участку аппарата, где жидкость и покидающий ступень пар находятся в равновесии. Число ТТ (nт), необходимое для получения дистиллята и остатка заданного состава, можно найти графически с помощью у-х-диаграммы (рис. 2), описывающей зависимость между равновесными молярными концентрациями паровой (у*) и жидкой (х) фаз. Для определения пт на графике строят (см. Массообмен) ступенчатую линию между равновесной кривой у* = (х) и ломаной линией AВС. Линия АВ отвечает ур-нию (3), линия ВС— ур-нию (2). В представленном примере для разделения исходной смеси на дистиллят состава xD и остаток состава xW требуется по 3 ТТ в укрепляющей и исчерпывающей частях колонны. Более точный метод расчета составов дистиллята и остатка при известном числе ТТ основан на последоват. вычислении составов пара и жидкости на каждой тарелке с использованием ур-ний рабочих линий и теплового баланса. Учитываются также изменения потоков по высоте колонны вследствие неравенства теплот испарения компонентов.
Из рис. 2 следует, что пт определяется положением рабочих линий в обеих частях колонны, к-рое, в свою очередь, зависит от R. На рис. 3 изображено неск. положений рабочих линий, однако существуют два предельных положения: первое-линии СВ для верха и AB для низа колонны, второе-линии СВ2 для верха и АВ2 для низа колонны. Первый предельный случай-бесконечно большое флегмовое число (R = ,; колонна работает "на себя", т. е. вся жидкость, полученная в результате полной конденсации паров в дефлегматоре, возвращается в колонну в виде флегмы; отбор дистиллята и выдача продукта не производятся, что в нормальных производств. условиях исключается; подобный режим удобен только для исследоват. целей), при этом рабочая линия совпадает с диагональю диаграммы. Отрезок b=xD(R+1), отсекаемый рабочей линией ABC на оси ординат, в соответствии с ур-нием (2) равен нулю и, следовательно, изменение рабочих концентраций отвечает ур-нию у = х, т. е. составы пара и жидкости равны в каждом сечении колонны. Второй предельный случай-рабочие линии пересекают равновесную кривую у*=(х) в точке В2. Ректификация возможна, но поскольку в точке В2 движущая сила равна нулю (у* = у), для проведения процесса потребуется колонна с бесконечно большой пов-стью фазового контакта, работающая при миним. флегмовом числе, к-рое составляет:
![]()
где у*-состав пара, равновесный с хр.
Положение рабочих линий СВ1 и B1A соответствует эксплуатации колонн в производств, условиях. Точка В1 может приближаться к верх. пределу В2 либо к ниж. пределу В; при этом флегмовое число, при к-ром функционирует колонна, или рабочее флегмовое число (Rраб), изменяется от Rмин до R = ,. С уменьшением R снижается расход теплоты на испарение жидкости в кубе колонны, однако уменьшается движущая сила, что приводит к необходимости увеличивать высоту колонны, т.е. к росту капитальных затрат. Оптим. флегмовое число следует определять на основе техн.-эконо-мич. расчетов. С известным приближением Roпт можно найти графически, исходя из зависимости затрат 3 на ректификацию от R (рис. 4). Эксплуатац. расходы увеличиваются прямо пропорционально R; кривая капитальных затрат имеет минимум, т.к. с возрастанием R уменьшается высота колонны, но увеличивается ее сечение; кривая суммарных затрат на ректификацию также имеет минимум, к-рый отвечает Roпт.
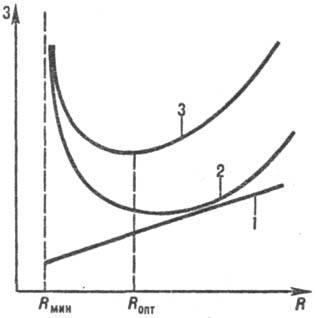
Рис. 4. К определению оптимального флегмового числа: 1-эксплуатац. расходы; 2-капитальные затраты; 3-общие затраты на ректификацию.
Выбор рабочего флегмового числа часто проводят приближенно по ф-ле: Rраб = Rмин, где = Rраб/Rмин -коэф. избытка флегмы (обычно не превышает 1,05-1,5). При отсутствии данных о для разделяемых смесей можно применять эмпирич. зависимость: R = 1,3Rмин + 0,3. Более точный метод расчета Rопт предполагает знание приведенных затрат и учет расходов, связанных с подачей сырья и подводом теплоты в колонну и организацией ее орошения, а также стоимость колонны и вспомогат. оборудования.
Равновесие между паром и жидкостью на реальных тарелках, как правило, не достигается и для определения их эффективности используют понятие кпд тарелки, или кпд Мерфри:
![]()
где
уn,
уп-1
-средние составы паров,
поднимающегося с n-й тарелки
и поступающего на нее;![]() -составпара,
равновесный с составом жидкости,
покидающей n-ю тарелку.
Коэф.
зависит от структуры
потоков
на тарелке,
физ.-хим. св-в смеси, конструкции
контактного устройства и изменяется в
пределах 0,3-0,9 (для одной ТТ
= 1). Величина
м.б. найдена по диффузионной модели
массообмена
между паром
и жидкостью,
если для данных типа тарелки
и смеси известно число единиц переноса
(Noy).
-составпара,
равновесный с составом жидкости,
покидающей n-ю тарелку.
Коэф.
зависит от структуры
потоков
на тарелке,
физ.-хим. св-в смеси, конструкции
контактного устройства и изменяется в
пределах 0,3-0,9 (для одной ТТ
= 1). Величина
м.б. найдена по диффузионной модели
массообмена
между паром
и жидкостью,
если для данных типа тарелки
и смеси известно число единиц переноса
(Noy).
При расчете насадочных аппаратов (обычно графически или аналитически) определяют число ТТ, необходимых для заданного разделения, и высоту насадки, эквивалентную по эффективности одной ТТ (ВЭТТ). Последнюю находят, как правило, по опытным данным или эмпирич. ур-ниям. Более строгий метод расчета основан на использовании ур-ний массо- и теплопереноса. В последнее время было установлено, что перенос ЛЛК из жидкости в пар связан как с диффузией, так и с теплообменом между паром и жидкостью. В любом сечении колонны т-ра пара выше т-ры жидкости, поэтому вследствие воздействия теплового пото-ка часть жидкости испаряется и примерно такое же кол-во пара конденсируется. Содержание ЛЛК в образующемся паре, естественно, выше, чем в жидкости, а содержание в ней ЛЛК после конденсации пара ниже, чем в паровой фазе. Т. обр., в результате испарения и конденсации возникает дополнит. конвективный поток ЛЛК из жидкости в пар за счет термической ректификации.
Общее кол-во ЛЛК, передаваемого из пара в жидкость, определяется суммой диффузионного (Nд) и термич. (Nтp) потоков. Поэтому локальный общий коэф. массопередачи К при ректификации находится по ур-нию:
![]()
где Каб = (1/у + m/х)-коэф. массопередачи, вычисляемый по ур-ниям физ. абсорбции; у, х-коэф. массоотдачи в паровой и жидкой фазах; m-наклон линии равновесия; ж-коэф. теплоотдачи при конденсации пара; r-теплота испарения; xi, yi-концентрации ЛЛК на границе раздела фаз; ti, tж-т-ры на границе раздела и в ядре потока жидкости. Средний коэф. массопередачи в пределах изменения концентраций в колонне от y1 до у2 рассчитывают по ур-нию:
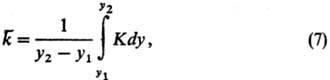
а высоту слоя насадкив колонне по ф-ле:

где G- расход пара;а-пов-сть контакта фаз в расчете на единицу объема аппарата; F-площадь поперечного сечения колонны. В общем случае потоки пара и жидкости, а также величины К и (у* — у) зависят от у и кривизны равновесной линии и вычисление H необходимо проводить численным методом или с помощью ЭВМ.
Периодическая ректификация (рис. 5). В куб колонны загружают определенное кол-во подлежащей разделению смеси, где она нагревается до т-ры кипения и испаряется. Образующиеся пары проходят через колонну, взаимодействуя в противотоке с жидкостью, поступающей из дефлегматора. В нем конденсируются выходящие из колонны богатые ЛЛК пары, направляемые далее в делитель потоков. Часть конденсата (флегма) поступает обратно в колонну, др. часть (дистиллят)-через холодильник в один из сборников.
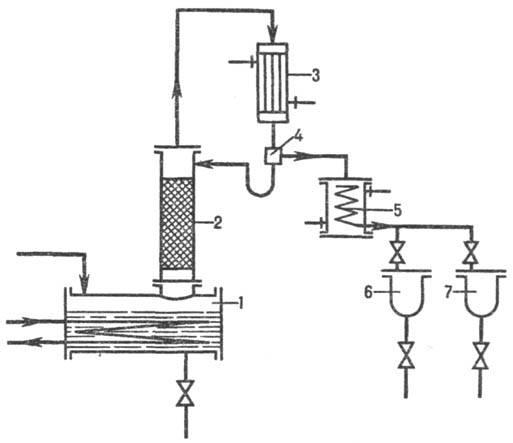
Рис. 5. Ректификационная установка периодич. действия: 1 -куб; 2-колонна; 3-дефлегматор; 4-делитель потоков; 5-холодильник; 6, 7-сборники.
Установки для периодической ректификации подразделяются на агрегаты, работающие при постоянном флегмовом числе, и агрегаты, функционирующие при переменном R и постоянном составе дистиллята. Наиб. широко распространены в пром-сти установки первого типа. Для этого случая изменение составов кубовой жидкости и дистиллята показано на рис. 6. В начале ректификации концентрация ЛЛК в кубовой жидкости равна xF, а в дистилляте-xD. По мере течения процесса концентрация ЛЛК в кубе будет уменьшаться и принимать значения x1, x2 и т. д. Соотв. понизятся концентрации ЛЛК в дистилляте, принимая значения x'D, х''D и т.д. В результате будет получен дистиллят среднего состава в пределах xD-xDW и остаток в кубе состава xW. Из рис. 6 следует, что при R.= const наклон всех рабочих линий, равный R/(R—l), не зависит от концентрации ЛЛК, поэтому линии смещаются параллельно своему первонач. положению. Средний состав полученного дистиллята м. б. рассчитан по ур-нию:
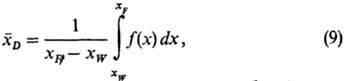
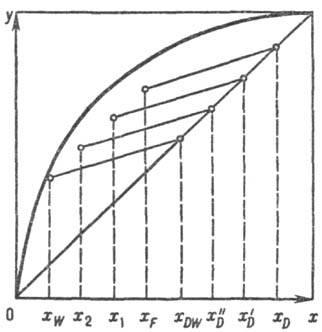
Рис. 6. Рабочие линии периодической ректификации с постоянным флегмовым числом.
если известна зависимостьмежду составами кубовой жидкости и дистиллята. При постоянном составе дистиллята, переменном R и известной его зависимости от концентрации ЛЛК графич. интегрированием можно найти среднее флег-мовое число по ур-нию:
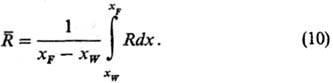
Азеотропная ректификация. Для нек-рых бинарных смесей кривые равновесия у* = (x) при определенных условиях пересекают диагональ y-x-диаграммы; в точке пересечения составы пара и жидкости одинаковы (азеотропная смесь), вследствие чего подобные смеси обычной ректификацией разделить невозможно. Поэтому к исходной смеси добавляют р-ри-тель-т.наз. разделяющий агент, образующий с одним из компонентов азеотропную смесь, к-рая при ректификации выделяется в виде дистиллята; кубовая жидкость представляет собой высококипящий компонент с миним. содержанием разделяющего агента. Однако его выделение из азеотропной смеси (дистиллята) затруднено. Один из методов, позволяющий осуществить рецикл р-рителя, заключается в применении таких разделяющих агентов, к-рые обладают ограниченной взаимной р-римостью в компонентах, отбираемых в виде дистиллята. При этом благодаря его расслаиванию в разделит. сосуде слой, обогащенный ЛЛК, поступает в среднюю часть регенерац. колонны, откуда в результате ректификации в виде кубового продукта отбирается ЛЛК исходной смеси, а в виде дистиллята-азеотроп, направляемый в разделит. сосуд (рис. 7).
Экстрактивную ректификацию используют обычно для разделения смесей близкокипящих компонентов, характеризующихся низкой относит. летучестью а. Разделение таких смесей приходится проводить в колоннах с очень большим числом ТТ и высоким расходом пара из-за необходимости поддерживать большое R. Однако если к исходной смеси добавить практически нелетучий разделяющий компонент, способный повысить осн. компонентов, разделение можно осуществить в двух ректификац. колоннах. На одну из верх. тарелок первой колонны подается разделяющий агент, к-рый раств. в потоке флегмы и повышает а смеси, в результате чего в виде дистиллята выделяется ЛЛК, а в виде остатка-смесь ТЛК и разделяющего агента. Эта смесь направляется в середину второй колонны, где, в свою очередь, разделяется на ТЛК (дистиллят) и остаток (разделяющий агент), к-рый возвращается в первую колонну.
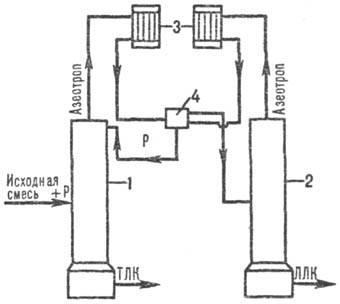
Рис. 7. Установка для азеотропной ректификации: 1 -ректификац. колонна; 2-регенерац. колонна; 3-дефлегматор; 4-разделит. сосуд; Р-разделяющий агент.
Методы азеотропной и экстрактивной ректификации находят широкое применение для разделения близкокипящих углеводородов нефти и сжиженных прир. газов, жидких смесей в произ-ве жирных к-т, получения безводного этилового спирта и др.
Молекулярная ректификация, наз. также многоступенчатой мол. дистилляцией, используется для разделения смесей малолетучих и термически нестойких в-в. Среди разл. конструкций аппаратов для такой ректификации практич. интерес представляют аппараты лестничного типа, принцип работы к-рых показан на рис. 8. Пары, испаряясь с пов-сти жидкости, напр. на ступени А3, конденсируются на наклонной пов-сти В3, а образующийся конденсат стекает в смежную вышестоящую ступень А4. Избыточное кол-во жидкости из ступени А4 переливается в ступень А3 и т.д., т.е. в аппарате осуществляется противоток жидкости и пара.
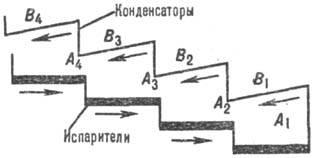
Рис. 8. Установка для молеку лярной ректификации.
Молекулярная ректификация реализуется в условиях высокого вакуума (неравновесного испарения), когда б. ч. испарившихся молекул без столкновения между собой долетает до пов-сти конденсации и остается на ней. При расчете разделения в описанном аппарате можно применить аналит. и графич. методы, используемые для тарельчатых аппаратов. При этом вместо коэф. относит. летучести, кпд тарелки и числа ТТ необходимо применять соотв. коэф. разделения при неравновесном испарении (н), кпд отдельной ступени и число теоретич. мол.тарелок:
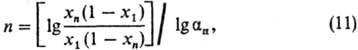
где n-число ступеней;
![]()
x1,
хn-концентрации
ЛЛК в первой и последней ступенях;![]() давления
паров
чистых компонентов 1 и 2; М1,
М2-их
мол. массы.
давления
паров
чистых компонентов 1 и 2; М1,
М2-их
мол. массы.
Ректификация многокомпонентных смесей Методы расчета. Из-за недостаточности данных о кинетике и механизме разделения многокомпонентных смесей для них нельзя задать полные составы продуктов разделения и поэтому первоначально принятые составы приходится корректировать. Без учета изменения кол-в потоков по высоте колонны вследствие разл. теплот испарения отдельных компонентов материальные балансы относительно известных составов представляются на каждой тарелке ур-нием:
![]()
где L, V-потоки жидкости и пара; xi+1,k, xi,k-составы жидкости по k-му компоненту, поступающей на ню тарелку и выходящей с нее; yi,k, yi-1,k-составы пара, выходящего с 1-й тарелки и поступающего на нее. При учете теплот испарения ур-ния балансов будут нелинейны:
![]()
Ур-ния (12) и (13) составляют мат. описание ректификации многокомпонентных смесей в противоточной тарельчатой колонне. С помощью этих ур-ний можно вычислять расходы и составы материальных потоков для всех тарелок колонны. Для расчета необходимо определить содержание осн. ЛЛК и ТЛК и примесей. На начальном этапе расчета содержанием каждого компонента в дистилляте приходится задаваться, и полный состав всех продуктов и состав кубовой жидкости находятся весьма приближенно.
Расходы материальных потоков и их составы на каждой ступени определяются последоват. расчетом от тарелки к тарелке. Существует много методов расчета, один из них-метод встречных вычислений, при к-ром исчерпывающая часть колонны рассчитывается в направлении от куба к тарелке питания (на нее поступает исходная смесь), а укрепляющая часть-к ней от дефлегматора. Несоответствие результатов вычислений составов продуктов на тарелке питания, полученных при расчете в двух направлениях, служит основанием для коррекции заданного первоначально состава продуктов. Далее задаются новым составом продуктов и определяют составы осн. компонентов на тарелке питания и т.д., пока составы на ней не станут одинаковы. Если в основе расчета лежит понятие о ТТ, предполагается, что состав пара на каждой тарелке равновесен составу жидкости; при использовании мат. модели на базе реальных тарелок для расчета составов пара, покидающего каждую тарелку, применяют ур-ние:
![]()
где Ei,k-кпд 1-й тарелки по k-му компоненту.
Независимое определение Ei по каждому компоненту не учитывает влияния движущих сил для разделения остальных компонентов, что может нарушить соотношение:
![]()
Поэтому при ректификации разделит. способность колонн необходимо определять на основе общих кинетич. закономерностей диффузии в многокомпонентных смесях (см. Диффузия). С помощью этих методов для паровой и жидкой фаз можно найти матрицу общих коэф. массопередачи, применяя правило аддитивности фазовых сопротивлений:
![]()
где [m]-матрица угловых коэф. касательных к равновесной линии для многокомпонентной смеси; [y], [x]-матрицы коэф. массоотдачи для паровой и жидкой фаз. Матрицу [К] можно использовать далее для решения ур-ния (14).
Синтез технологических схем разделения. Для разделения n-компонентной смеси требуется п — 1 колонн, однако число возможных вариантов технол. схем с расчетом числа продуктов и способов их получения увеличивается экспоненциально. На рис. 9 показано, что для разделения смеси компонентов ABCD, расположенных в порядке возрастания т-р кипения, возможны 5 вариантов схем деления; для смеси из 10 компонентов число возможных схем составляет 4862.
Синтез оптим. схем разделения-сложная проблема теории ректификации. Постановка задачи включает перечень продуктов разделения и требования к ним по составу целевых компонентов и примесей. При синтезе наиб. оптимальной схемы разделения вначале нужно провести анализ физ.-хим. св-в компонентов исходной смеси, условий фазового равновесия в многокомпонентной системе, теплового баланса схемы, позволяющей выявить потенц. источники и стоки энергии, и затем приступить к созданию вариантов схемы разделения.
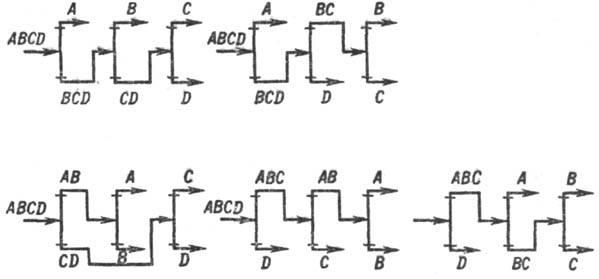
Рис. 9. Возможные варианты схем разделения компонентов смеси ABCD.
На основе анализа фазовых равновесий выясняется принципиальная возможность разделения и выявляются ограничения, обусловленные, напр., образованием азеотропов и наличием близкокипящих компонентов. В этом случае возникает необходимость применения азеотропной или экстрактивной ректификации. Эксплуатац. затраты, связанные с расходом энергии, могут достигать при ректификации 70% общей стоимости разделения, поэтому при проектировании надо решать задачу рационального сочетания флегмового числа, от к-рого зависит расход энергии, и числа ТТ, т.е. высоты колонны, определяющей капитальные затраты. Оптим. схема разделения должна отвечать минимуму затрат. При выборе схемы, состоящей из ряда колонн, снижение энергетич. затрат возможно за счет рекуперации тепловых потоков благодаря различию т-р кипения продуктов разделения (напр., высококипящие компоненты можно использовать для подогрева низкокипящих).
Большая экономия энергии м. б. достигнута путем применения схемы с тепловым насосом (рис. 10). В данном случае пары дистиллята, выходящие из колонны, сжимаются компрессором до давления, соответствующего требуемой т-ре его конденсации в кубе колонны; при этом отпадает необходимость в дефлегматоре и сокращаются расходы пара и воды. С целью экономии капитальных затрат иногда выгодно использовать вместо неск. простых колонн одну сложную колонну с отпарными секциями (см. ниже) и боковыми отборами отдельных фракций. Такие колонны используют при разделении углеводородных газов и переработке нефти.
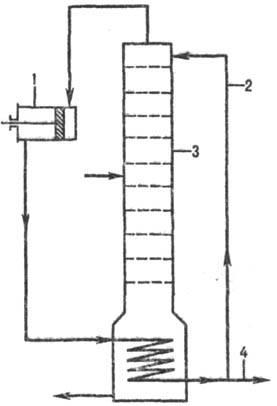
Рис. 10. Установка с тепловым насосом: 1-колонна; 2-компрессор; 3— флегма; 4-дистиллят.
Типичный пример сложной колонны-колонна атм. перегонки обезвоженной и обессоленной нефти (рис. 11); продукты разделения: газ, состоящий из углеводородов С2-С4; фракция бензина до 180 °С; фракция керосина 160-250 °С;
фракции легкого (220-300 °С) и тяжелого (280-350 °С) дизельных топлив; фракция атм. газойля 350-380 °C; мазут (фракция выше 350-380 °С). Каждый боковой погон из осн. колонны направляется в спец. колонны (секции), где происходит отпарка из него легких фракций. Из отпарных колонн пары возвращаются в осн. колонну, а жидкость отбирается
в виде целевых фракций. Ректификацию используют, наряду с указанными выше областями применения, при получении азота и кислорода из воздуха (см. Воздуха разделение), ряда чистых металлов, тяжелой воды, в пром-сти орг. синтеза и др. В лаб. практике применяют в осн. те же, что и в пром-сти, способы ректификации, проводимой в стеклянных насадочных либо тарельчатых ректификац. колонках; периодическую ректификацию используют для разделения небольших кол-в (неск. десятков г), непрерывную-значит. кол-в (сотни и тысячи г) смесей.
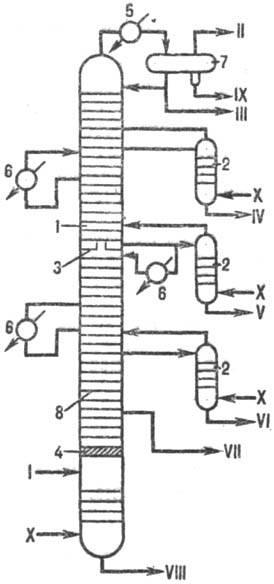
Рис. 11. Установка атмосферной перегонки нефти: 1-осн. колонна; 2-отпарные колонны (секции); 3-тарелка питания; 4-отбойник; 5-конденсатор; 6-холодильник; 7-емкость орошения; 8-рабочие тарелки; I-нефть; II - газ; III -бензин; IV-керосин; V, VI-легкое и тяжелое дизельные топлива; VII-атм. газойль; VIII-мазут; IX—во да; Х-водяной пар.
Ректификация-один из самых энергоемких хим.-технол. процессов. Поэтому в хим. произ-вах все чаще применяют альтернативные процессы и методы разделения. К ним относят: испарение через мембрану (см. Мембранные процессы разделения), осуществляемое в аппаратах пленочного типа; противоточную кристаллизацию с непрерывным массообменом (экономия энергии достигается благодаря тому, что теплота плавления разделяемых в-в, как правило, существенно меньше, чем теплоты их парообразования; см. также Кристаллизационные методы разделения смесей)и др. Однако, несмотря на все большее распространение этих и иных альтернативных процессов и методов, ректификация по-прежнему сохраняет свое значение в хим. отраслях пром-сти, особенно в нефтепереработке и нефтехимии.
Отличие ректификации от перегонки.
Сущность процесса ректификации сводится к выделению из смеси нескольких или, в нашем случае, одной жидкости с отличной от других температурой кипения в более или менее чистом виде. Это достигается нагреванием и испарением такой смеси с последующим многократным тепло- и массообменном между жидкой и паровой фазами; в результате часть легколетучего компонента переходит из жидкой фазы в паровую, а часть менее летучего компонента—из паровой фазы в жидкую. Важно! Ректификация позволяет получить чистый компонент без примесей, в отличие от обычной дистилляции (перегонки), когда на выходе мы имеем не контролируемый набор различных веществ.
В чем заключается принципиальное отличие процессов изотермической и изогидрической кристаллизации.
ИЗОТЕРМИЧЕСКАЯ КРИСТАЛЛИЗАЦИЯ
Перевод исходного раствора, характеризуемого точкой А (см. рис. 1), в пересыщенное состояние можно осуществить и за счет частичного удаления растворителя при выпаривании раствора. Такой метод получил название изотермической кристаллизации, так как выпаривание насыщенного раствора происходит при постоянной температуре его кипения.
Изменение концентрации исходного ненасыщенного раствора при выпаривании изобразится линией AEG, которая показывает, что с повышением концентрации раствора соответственно возрастает и его температура кипения. Только после перехода раствора в насыщенное состояние при концентрацииС 1 " (точкаЕ) температура уже больше не меняется и остается равнойt 1 "".
Понятно, что приведенное выше изображение процесса кристаллизации по линии AEG на диаграмме растворимости является условным, и точкаG характеризует лишь общее пересыщение раствора, которое может быть получено при удалении из него определенной части растворителя. В большинстве случаев кристаллизация раствора при выпаривании протекает при постоянной концентрации, очень близкой к состоянию насыщения для данной температурыt 1 "" .
 Выбор
того или иного метода кристаллизации
зависит, в первую очередь, от характера
изменения растворимости вещества при
различной температуре. Для солей,
растворимость которых резко уменьшается
с понижением температуры, целесообразной
является изогидрическая кристаллизация.
В этом случае даже при сравнительно
небольшом охлаждении раствора из него
будет выделяться значительное количество
соли (см. кривую растворимости KNO3 на
рис. 2). Именно изогидрической кристаллизацией
получают большинство солей с резко
выраженной прямой растворимостью (NaNO3 , К2 Сr2 О7 , NH4 C1, CuS04 -5H2 0 и др.).
Выбор
того или иного метода кристаллизации
зависит, в первую очередь, от характера
изменения растворимости вещества при
различной температуре. Для солей,
растворимость которых резко уменьшается
с понижением температуры, целесообразной
является изогидрическая кристаллизация.
В этом случае даже при сравнительно
небольшом охлаждении раствора из него
будет выделяться значительное количество
соли (см. кривую растворимости KNO3 на
рис. 2). Именно изогидрической кристаллизацией
получают большинство солей с резко
выраженной прямой растворимостью (NaNO3 , К2 Сr2 О7 , NH4 C1, CuS04 -5H2 0 и др.).
Рис. 2. Кривые растворимости в воде КNО 3 (1), Ва(NО3 )2 (2) и NaCl (3).
В тех случаях, когда растворимость соли почти не меняется при изменении температуры, кристаллизация охлаждением становится неэффективной и применяется изотермическая кристаллизация Кривая растворимости, например NaCl (см. рис.2) показывает, что при охлаждении насыщенного раствора из него выпадает лишь очень небольшое количество соли, поэтому кристаллизация NaCI проводится всегда выпариванием.
Изотермическая кристаллизация применяется также для солей с обратной растворимостью, например для Na 2 S04 , растворимость которого, начиная с 32,4° С, уменьшается с повышением температуры. Для кристаллизации солей с резко выраженной обратной растворимостью иногда используют просто нагревание раствора до высоких температур. Так, например, получают безводный кристаллический сульфит натрия Nа2 S0з и сульфат марганца MnS04 .
На практике в ряде случаев комбинируют рассмотренные выше методы создания пересыщения. Так, при вакуум-кристаллизации раствор охлаждается за счет адиабатического испарения части растворителя. Этот метод кристаллизации особенно эффективен для солей, растворимость которых сравнительно плавно уменьшается с понижением температуры, например для КС1 (см. рис. 2), (NH 4 )2 S04 , FeS04 -7H2 0 и др.
Частичное испарение растворителя характерно и для некоторых кристаллизаторов охладительного типа, например, для башенных кристаллизаторов, барабанных с воздушным охлаждением, качающихся и др.
ИЗОГИДРИЧЕСКАЯ КРИСТАЛИЗАЦИЯ
Для осуществления процесса кристаллизации в растворе необходимо создать пересыщение. По способам его создания различают два основных метода кристаллизации: 1) охлаждение горячих насыщенных растворов (изогидрическая или политермическая кристаллизация) и 2) удаление части растворителя путем выпаривания (изотермическая кристаллизация). ПОЛИТЕРМИЧЕСКАЯ КРИСТАЛЛИЗАЦИЯ Растворимость большинства веществ уменьшается с понижением температуры. Поэтому при охлаждении горячих растворов возникает пересыщение, обусловливающее выделение кристаллов. Этот метод также получил название изогидрической кристаллизации, поскольку при его осуществлении количество растворителя (например, воды) остается постоянным. Рис. 1. К пояснению методов кристаллизации (см. текст). На диаграмме растворимости (рис. 1) охлаждение горячего ненасыщенного раствора, имеющего температуру t1 и концентрацию C1 (точка А), до конечной температуры t2 условно можно изобразить линией АС, которая пересекает кривую растворимости в точке В, характеризующей насыщенное состояние раствора при температуре t1'. Если кристаллизация раствора начинается только после его охлаждения до температуры t2, при которой и заканчивается полное снятие пересыщения, то процесс кристаллизации изобразится линией CD, а конечное состояние раствора-точкой D на кривой растворимости, соответствующей равновесной концентрации C2. В том случае, если раствор не способен к образованию сколько-нибудь заметного пересыщения, процесс его охлаждения и кристаллизации изобразится линией АВD. В зависимости от скорости охлаждения раствора и его способности образовывать пересыщение реальный процесс может протекать также по линиям AB'D'D или AB"D.\
Политермическая кристаллизациясвязана с охлаждением растворов и основана на различной растворимости веществ в растворе в зависимости от температуры. Иногда используют метод политермической выпарки. [1]
Политермическая кристаллизацияосуществляется охлаждением насыщенных растворов и применяется для веществ, растворимость которых при повышенных температурах заметно выше, чем при низких. [2]
Политермическая кристаллизацияосуществляется охлаждением насыщенных растворов и применяется для веществ, растворимость которых при повышенных температурах выше, чем при низких. [3]
Политермическая кристаллизация, основанная на различной растворимости совместно присутствующих солей в зависимости от температуры. Примером такого процесса может служить выделение хлористого калия из сильвинита ( стр. [4]
При политермической кристаллизациипересыщенный раствор образуется за счет охлаждения систе - мы. Этот процесс протекает при переменной температуре
Процесс политермической кристаллизациив случае отсутствия взаимодействия между компонентами системы рассмотрен ниже. Процессы, в которых принимают участие кристаллогидраты и двойные соли, будут рассмотрены при описании применения физико-химического анализа в технологии некоторых продуктов, получаемых из галургического сырья. [6]
Сущность политермической кристаллизациизаключается в том, что готовят концентрированный раствор при повышенной температуре, затем его охлаждают до комнатной или до более низкой температуры. Чем больше разность температуры, тем больше образуется кристаллов. [7]
В единичном реакторе полного смешения концентрация исходных реагентов невелика, так как мгновенно падает до конечного значения. Поэтому в нем малы скорость химического превращения и степень превращения. Для повышения этих показателей применяют ряд последовательно расположенных реакторов полного смешения - каскад реакторов. Концентрация исходных реагентов СА в такой системе изменяется ступенчато. При этом изменение концентрации происходит мгновенно при входе реакционной смеси в каждый реактор.
В некоторых случаях процесс химического превращения вещества проводится не в одном аппарате смешения, а в нескольких таких аппаратах, соединенных последовательно (рис. 6.6,г). Такая система, состоящая в некоторых случаях из 20 и более аппаратов, получиланазвание каскада реакторов (батареи реакторов). В каскаде реакторов изменение концентрации реагирующих веществ носит ступенчатый характер (рис. 6.5,в), так как продукт реакции предыдущего аппарата является исходным реагирующим веществом в последующем аппарате.
Гидродинамический режим работы каскада реакторов является промежуточным и зависит от числа аппаратов: с увеличением числа реакторов в каскаде он приближается к режиму вытеснения, а при уменьшении к режиму смешения. В каскаде увеличивается время пребывания реагирующих веществ по сравнению с одним реактором смешения, а также растет выход продуктов реакции по сравнению с реактором вытеснения.
Ректификация (от лат. rectus — прямой и facio — делаю) — это процесс разделения бинарных или многокомпонентных смесей за счет противоточного массо- и теплообмена между паром и жидкостью. Ректификацию можно проводить периодически или непрерывно. Ректификацию проводят в башенных колонных аппаратах, снабженных контактными устройствами (тарелками или насадкой) ректификационных колоннах.
Ректификацию широко применяют в промышленности, например для получения ректификованного этилового спирта, с отделением сивушных масел и альдегидных фракций, для выделения бензинов, керосинов и других фракций из нефти, а также получения компонентов воздуха (кислорода, азота, инертных газов).
Разделение путем перегонки основано на различной температуре кипения отдельных веществ, входящих в состав смеси. Так, если смесь состоит из двух компонентов, то при испарении компонент с более низкой температурой кипения (низко- кипящий компонент) переходит в пары, а компонент с более высокой температурой кипения (высококипящий компонент) остается в жидком состоянии. Полученные пары конденсируются, образуя дистиллят или ректификат, а неиспаренная жидкость называется остатком.
В результате перегонки низкокипящий компонент переходит в дистиллят, а высококипящий – в остаток. Такой процесс называется простой перегонкой. При этом не достигается полного разделения смеси. Оба компонента являются летучими, оба переходят в пары, но в разной степени. Поэтому образующиеся при перегонке пары не представляют собой чистого низкокипящего компонента.
Из-за большой летучести низкокипящий компонент испаряется в большей степени, чем высококипящий компонент. Значит, в дистилляте содержание низкокипящего компонента выше, чем в исходной смеси, а в остатке наоборот: содержание низкокипящего компонента ниже, чем в исходной смеси. В этом и является отличие перегонки от выпаривания (при выпаривании растворенное вещество нелетучее, а в пары переходит только летучий компонент).
Простую перегонку применяют для грубого разделения смесей или для предварительной очистки продуктов от нежелательных примесей.
Для достижения наиболее полного разделения компонентов применяют достаточно сложный вид перегонки – ректификацию.
Ректификация заключается в многократном испарении исходной смеси и конденсации образующихся паров, в противо- точном воздействии паров, образующихся при перегонке, с жидкостью, получаемой при конденсации паров.
Способ разделения смеси на компоненты путем ректификации является основным в спиртовом и ликеро-водочном производствах, в производстве эфирных масел, при переработке нефтепродуктов и др.
Общий вид аппаратов для процессов с участием твердой и жидкой фаз.
Экстракторы периодического и полунепрерывного действия. наиб. распространены камерные аппараты (реакторы) с мех., пневматич. или пневмомех.перемешиванием, а также т. наз. настойные чаны с неподвижным слоем твердых частиц с циркуляцией (перколяторы) и без циркуляции экстрагента. Аппараты для экстрагирования в плотном слое обычно располагаются вертикально и имеют комбинир. форму: в осн. части цилиндрическую, с одного или обоих концов - форму усеченного конуса (рис. 1, а). На решетку сверху загружается слой твердого материала, через к-рый сверху вниз протекает экстрагент; для выгрузки твердого остатка служит откидное днище.
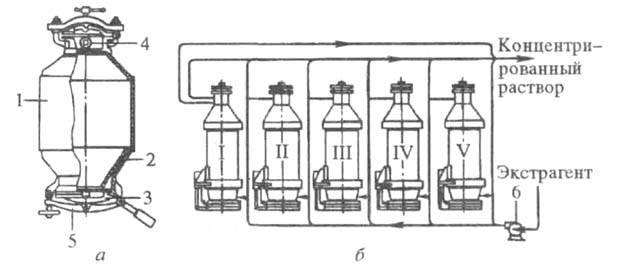
Рис. 1.Экстракторыпериодического действия: а - единичный аппарата; б -батарея аппаратов (I-V); 1 - корпус; 2 - ложное днище (решетка); 3 - откидное днище; 4 - штуцер для ввода свежего экстрагента; 5 - штуцер для отвода концентрированного р-ра; 6 -насос.
Последоват. соединение 4-16 таких аппаратов в батарею (рис. 1, б)позволяет перейти к полунепрерывной противоточ-ной схеме. Благодаря замкнутой системекоммуникаций удается периодически отключать от циркуля ц. системы один из аппаратов, освобождать его от полностью истощенного материала и заполнять свежим. Далее этот аппарат снова включают в систему циркуляции и подают в него наиб. обогащенный экстрагент, прошедший через все остальные аппараты; затем отключают след, аппарат, в к-рый до этого поступал чистый экстрагент, и т.д. С увеличением числа аппаратов процесс приближается к непрерывному. Гл. недостатки описанныхэкстракторов, к-рые продолжают широко применяться в хим. произ-вах: большие затраты ручного труда при их эксплуатации, значит. потери экстрагируемого в-ва при выгрузке, высокая металлоемкость, трудность регулирования работы.Экстракторыпериодич. действия используют в произ-ве небольших партий фармацевтич. препаратов, настоев, морсов и др.Экстракторыполунепрерывного действия (батарея аппаратов) малоэффективны, громоздки и сложны в обслуживании.
Экстракторы непрерывного действия. К осн.экстракторамотносятся шнековые и ленточные аппараты. Шнековы иэкстрактор(рис. 2) представляет собой трехколонный аппарат с транспортирующим органом шнекового типа. Твердая фаза последовательно перемещается через загрузочную, горизонтальную и экстракц. колонны навстречу движущемуся экстрагенту. В верх. части загрузочной колонны имеется сито для отделения экстракта от твердой фазы. Достоинства аппарата - малая металлоемкость и небольшая занимаемая площадь. Недостатки обусловлены конструкцией шнека, вокруг вала к-рого закручивается твердый материал; поэтому иногда шнек заменяют цепным транспортирующим органом. Ленточныйэкстрактор(рис. 3) имеет стальной корпус, внутри к-рого расположен транспортер с перфорир. лентой. Подаваемый в аппарат материал движется слоем высотой 0,6-1,2 м по верх. ветви транспортера.
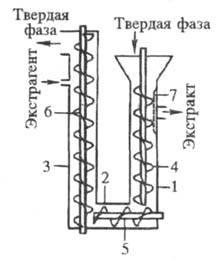
Рис. 2.Шнековыйэкстракторнепрерывного действия: 1, 2, 3 - загрузочная, горизонтальная и экстракц. колонны; 4-6 - шнеки; 7 -разделит. сито.
Для равномерного распределения экстрагента по пов-сти материала над слоем размещены распылители. Пройдя через слой материала, р-р поступает в воронку, откуда насосомподается в смежную зону, к-рая расположена в направлении, противоположном движению ленты. Распространены такжероторные аппаратыкарусельного типа, реализующие тот же принцип действия.
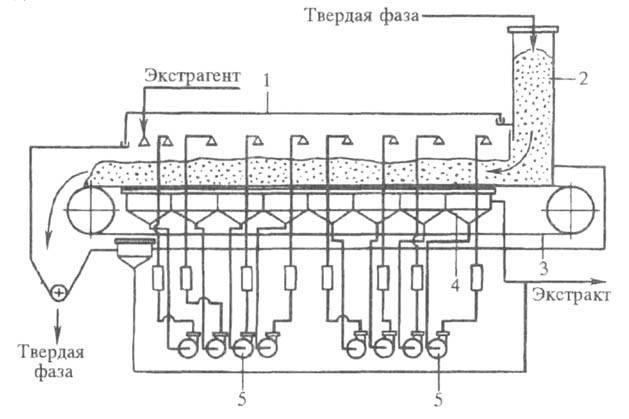
Рис. 3.Ленточныйэкстракторнепрерывного действия: 1 - корпус; 2 - бункер; 3 - ленточный транспортер; 4 - воронка; 5 -насосы.
Преимущества экстракторовнепрерывного действия, применяемых в многотоннажных произ-вах, перед периодически функционирующими аппаратами: более высокий коэф. массоотдачи от пов-сти твердых частиц к экстрагенту; полное исключение ручного труда при обслуживании; возможность создания экстрактов большой единичной мощности и автоматизации экстрагирования.
Интенсификация процесса
По сравнению с растворениемэкстрагирование протекает медленнее. Для его интенсификации целесообразны след. способы: 1. Повышение т-ры экстрагента. Приводит к увеличению коэф.диффузии, что ускоряет извлечение растворенного и твердого в-в; в последнем случае возрастает и движущая сила процесса cs— c1[см. ур-ние (8)]. При повышении т-ры снижается такжевязкостьэкстрагента, вследствие чего уменьшаются потери напора на прокачку р-рителя через слои извлекаемого в-ва. 2. Повышение относит. скорости движения фаз. Способствует увеличению коэф. массоотдачи, что сокращает время экстрагирования (если процесс не лимитируется внутр.диффузией). 3. Интенсивноеперемешивание. Приводит к обновлению пов-сти контакта твердых частиц с экстрагентом (эффективно при внешнедиффузионном сопротивлении). 4. Повышениедавления. Уменьшает объемвоздуха, "защемленного" в пористом объеме частиц при погружении твердого в-ва в экстрагент, и, Следовательно, восстанавливает нарушенный при этом контакт внутр. пов-сти частиц сжидкостью. 5. Подвод энергии (вибрации, пульсации, ультразвуковые и инфразвуковые колебания). Кроме того, при хим. р-циях между в-вом и экстрагентом процесс можно ускорить, повышаяконцентрациюизвлекаемого в-ва. Экстрагирование используют: а) для извлечения соед.редких металлов,урана,серыи др. изруд; б) для извлечения из пористых продуктовспеканияразл. в-в (произ-воглинозема, NaF и т.д.); в) для выделения орг. соед. из растит. сырья в произ-вахсахара, растит, иэфирных масел, р-римых кофе ичая, лек. ср-в и др.; г) для образования пористых структур путем добавления и послед. извлечения р-римого в-ва после фиксации структуры (напр., в произ-ве пористыхпластмасс, применяемых как изоляц. материал).
70-ый Возможно не правильно)
71 Барабанные кристаллизаторы
Барабанный вращающийся кристаллизатор с водяным или воздушным охлаждением является одним из наиболее распространенных механических кристаллизаторов. Аппарат с водяным охлаждением представляет собой вращающийся барабан, установленный под небольшим углом 50 0 (уклон 1:100- 1:200) к горизонту 5 10 об/мин. На барабане закреплены бандажи , опирающиеся на опорные ролики. Для предотвращения осевого смещения у одного бандажа устанавливаются упорные ролики. Вращение передается от привода через зубчатый венец, укрепленный на барабане. Горячий раствор подается в верхний конец барабана и медленно движется вдоль его оси, занимая по высоте сечения 1/8 -1/5 диаметра барабана (100 200 мм). Толщина слоя раствора в барабане, угол наклона и число оборотов барабана- факторы определяющие время пребывания раствора в аппарате, выбираются в зависимости от свойства раствора и требований к продукту. Как и во всех механических кристаллизаторах с водяным охлаждением, в барабанных аппаратах образуются довольно мелкие, но внутренне однородные кристаллы. Существенным недостатком таких аппаратов является значительная инкрустация поверхности барабана, громоздкость.
Для устранения пристенных осадков в барабан на всю его длину помещают тяжелую цепь, шарнирно закрепленную на его верх нем конце. При вращении барабана цепь перекатывается по его внутренней поверхности и сбивает наросты соли. В целях предупреждения образования инкрустаций используются барабанные кристаллизаторы с воздушным охлаждением. Раствор здесь охлаждается сильной струей воздуха, подаваемой вентилятором противотоком движению раствора со скоростью 4 5 м/с. Охлаждение происходит, главным образом, за счет испарения части растворителя.