

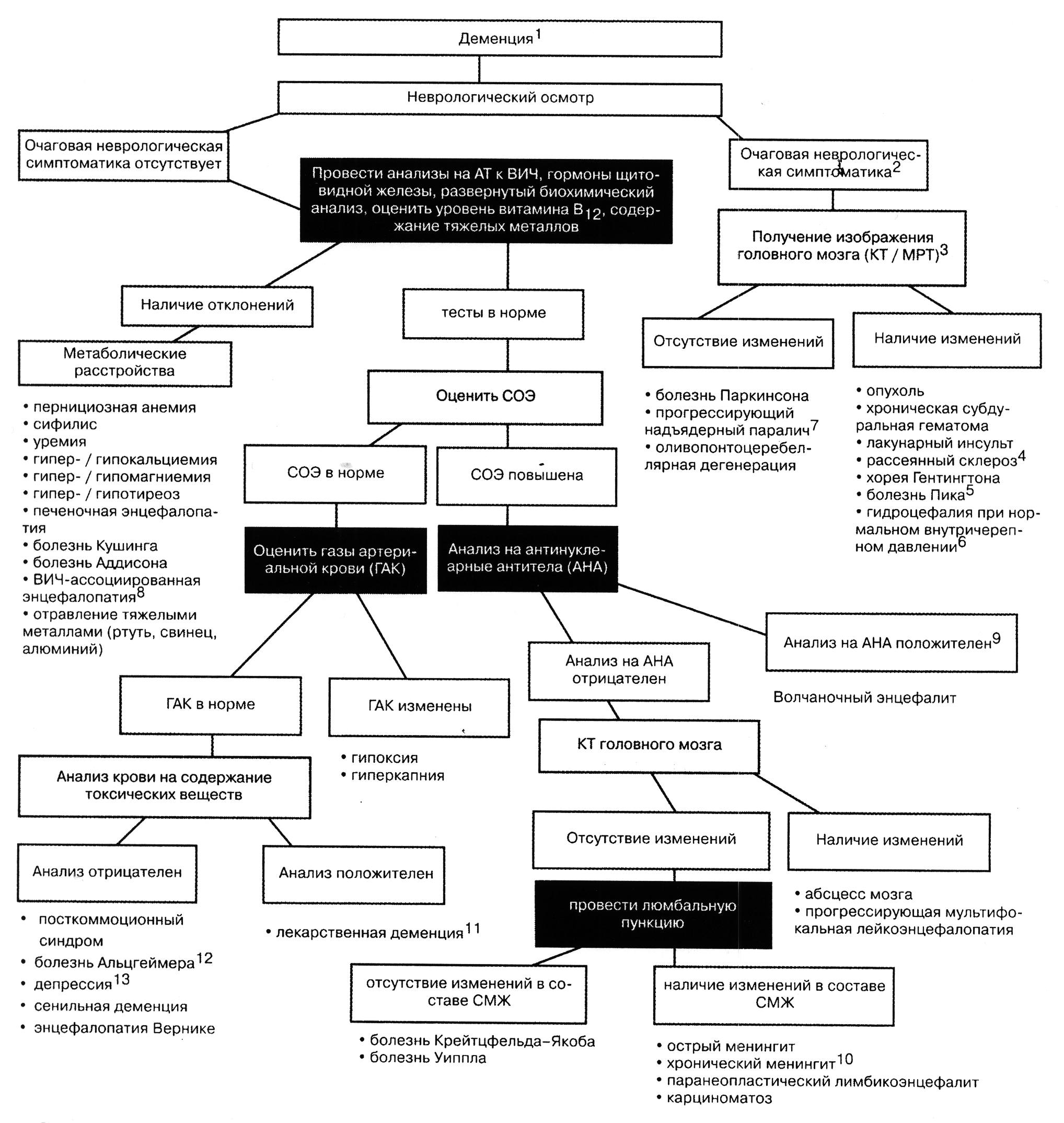
Алгоритм: «Деменция»
1. Деменция — приобретенное состояние, характеризующееся снижением умственных способностей. Деменция — не диагноз, а симптом какого-либо заболевания. Она может затрагивать память, речевые или познавательные способности, пространственную ориентацию, личность. Как правило, поражаются одновременно несколько функций. Деменция — это глобальное явление; ее следует отличать от очаговых дефектов, таких как афазия или амнезия. Деменцию необходимо также дифференцировать с острыми нарушениями сознания, которые длятся несколько часов или дней. Деменция поражает почти 15% людей в возрасте старше 65 лет и в меньшей степени — более молодых. Почти у 50% больных в основе деменции лежат метаболические или структурные нарушения, которые можно лечить.
2. Очаговые неврологические симптомы — это симптомы, появляющиеся при поражении одного из участков центральной нервной системы, при этом другие функции относительно сохранны. Очаговые симптомы в большинстве случаев касаются двигательных функций и чувствительности, но можно выявить и изменения памяти, равновесия, познавательных функций.
3. Современная техника позволяет получить изображение головного мозга различными способами. Выбор того или иного метода исследования зависит от предполагаемого диагноза. Так, компьютерная томография (КТ) стала стандартным исследованием для выявления структурных нарушений мозга и его желудочков. Магнитно-резонансная томография (МРТ) более надежна для обнаружения минимальных изменений белого и серого вещества мозга, как, например, при лакунарном инсульте и рассеянном склерозе. Новый метод — позитронно-эмиссионная томокрафия (ПЭТ) — может помочь в диагностике многих метаболических расстройств.
4. Рассеянный склероз — одна из причин ранней деменции. Деменция при рассеянном склерозе обычно сопровождается другими клиническими симптомами, характерными для поражения разных участков головного мозга, зрительного нерва и спинного мозга. Заболевание начинается в возрасте 15-50 лет. Чувствительным методом диагностики рассеянного склероза является МРТ, позволяющая визуализировать участки демиелинизации (хотя и не всегда).
5. Болезнь Пика часто путают с более распространенной болезнью Альцгеймера из-за сходных форм деменции — с преобладанием двигательных симптомов. Деменция при болезни Пика связана с дегенерацией височных и лобных долей. Ведущим симптомом может быть афазия. При КТ выявляется увеличение височных рогов и атрофия лобных долей. Болезнь Пика характеризуется быстрым прогрессированием; метода ее лечения - не существует.
6. Гидроцефалия при нормальном внутричерепном давлении - хроническое состояние, при котором происходит постепенное накопление спинномозговой жидкости (СМЖ) и увеличение желудочков мозга. Давление СМЖ в норме. Причина гидроцефалии при нормальном внутричерепном давлении неясна, так же как и ее клиническое значение. У многих пациентов с необструктивной (открытой) формой гидроцефалии имеются явления деменции, замедление психомоторных функций, неустойчивая походка и недержание мочи. В то же время признаки гидроцефалии выявляются при КТ почти у 5% лиц старше 60 лет, при этом у большинства таких пациентов признаков деменции нет. Данных о том, является ли подобная гидроцефалия обратимой после шунтирования желудочков, до настоящего времени нет.
7. Прогрессирующий надъядерный паралич (синдром Стила-Ричардсона-Ольшевского), напоминая болезнь Паркинсона, характеризуется менее выраженным тремором и наличием офтальмоплегии. Деменция развивается при длительном течении заболевания, и, по сравнению с деменцией при болезни Паркинсона, отличается более быстрым снижением умственных способностей. Эффективного лечения этого заболевания не существует.
8. Деменция при СПИД характеризуется изменениями сознания, мотойных функций и поведения. По-видимому, она является результатом непосредственного воздействия на клетки головного мозга вирусов иммунодефицита человека (ВИЧ). Такая деменция развивается постепенно, прогрессируя в течение многих месяцев или лет. По мере прогрессирования заболевания присоединяется слабость в руках и ногах, недержание мочи и кала. Иногда отмечаются тремор и миоклонус. Заболевание делает больных совершенно беспомощными. Эту форму деменции следует дифференцировать с другими - поддающимися лечению формами деменции при СПИД, обусловленными оппортунистическими инфекциями, воздействием лекарственных средств и депрессией.
9. К деменции может приводить острый энцефалит на фоне системной красной волчанки или других васкулитов. Точный механизм развития такой деменции неизвестен. Появление признаков деменции у пациентов с васкулитами требует интенсивного лечения.
10. Хронические формы менингита обычно вызываются латентными инфекциями. С возникновением разных форм деменции были связаны грибковые инфекции, особенно вызываемые видами Cryptococcus и Coccidioides. Деменция описана также при менингитах туберкулезной этиологии, саркоидозе и карциноматозе.
11. Химические вещества являются наиболее частой причиной деменции, причем вызывать ее могут как лекарственные средства, так и токсичные соединения. С развитием деменции связывают прием таких препаратов, как транквилизаторы (большие и малые), антидепрессанты, наркотические средства, антиконвульсанты, гипотензивные и антихолинергические препараты, дигоксин, а также некоторые антибиотики и противоопухолевые средства. Способность этих препаратов вызывать деменцию более заметна у пожилых больных. Ядовитые соединения, способные вызывать деменцию, — это тяжелые металлы, органические растворители, инсектициды.
12. Болезнь Альцгеймера — наиболее частая причина деменции. Болезнь Альцгеймера развивается постепенно; одним из наиболее ранних симптомов являются нарушения кратковременной памяти. Могут быть выражены эмоциональная лабильность и именная афазия. Основные двигательные и чувствительные функции на ранних стадиях болезни сохраняются, однако могут развиваться апраксия и недержание мочи/кала. Причина болезни Альцгеймера неизвестна; рассматривается роли вирусных инфекций, генетической предрасположенности, нейрохимических нарушений. При патоморфологическом исследовании в головном мозге обнаруживают сенильные бляшки, нейрофибриллярные узлы, грануловакуолярную дегенерацию нервных клеток. Заболевание постепенно прогрессирует и в итоге приводит к потере большинства когнитивных функций.
13. Депрессию иногда практически невозможно отличить от деменции. Особенно затруднена диагностика депрессии у пожилых лиц, которые часто предъявляют жалобы на тоску, беспомощность, бессонницу; потеря веса не всегда очевидна. Если никаких причин «деменции» выявить не удалось, необходимо тщательное психиатрическое обследование. Многих пациентов даже с очень глубокой депрессией можно успешно лечить, возвращая им нормальный психический статус.
Обследование пациента с деменцией
1. КТ/МРТ головы
2. Исследование крови: общий анализ, мочевина, электролиты, глюкоза, печеночные ферменты, кальций, гормоны щитовидной железы, белок и фракции, витамин Big. сифилис, ВИЧ, AHA
3. Рентгенография органов грудной полости
4. Электроэнцефалография
5. Анализ мочи на токсины, метаболиты лекарственных препаратов и соли тяжелых металлов
По специальным показаниям:
* люмбальная пункция
* биопсия головного мозга
* церебральная ангиография
Наиболее частые причины деменции:
* Болезнь Альцгеймера
* Сосудистая деменция (множественные лакунарные инфаркты, субкортикальная лейкоэнцефалопатия Бинсвангера)
* Алкоголизм*
* Болезнь Паркинсона
* Лекарственная интоксикация*
Менее частые причины:
* Дефицит витаминов (В12 - пернициозная анемия*, тиамина — энцефалопатия Вернике*, никотиновой кислоты - пеллагра*)
* Эндокринные заболевания: гипотиреоз*, надпочечниковая недостаточность, синдром Кушинга*, гипо- и гиперпаратиреоз*
* Органная недостаточность: почечная недостаточность*, печеночная недостаточность*, дыхательная недостаточность*
* Хронические инфекции: ВИЧ, нейросифилис*, паповавирусы (прогрессирующая мультифокальная лейкоэнцефалопатия), прионы (болезнь Крейтцфельда-Якоба и Герстманна-Штросслера), туберкулез*, грибковые* и паразитарные поражения* ЦНС, саркоидоз*, болезнь Уиппла*
* Травма и диффузное поражение головного мозга: посттравматическая деменция, хроническая субдуральная гематома*, постаноксическая энцефалопатия, постэнцефалитная энцефалопатия, гидроцефалия при нормальном внутричерепном давлении*
* Онкологические заболевания: первичные опухоли головного мозга*, метастазы в головной мозг*, паранеопластический лимбический энцефалит.
Токсические поражения: лекарственные и наркотические препараты*, отравление солями тяжелых металлов*, диализная деменция, органические токсины * Психические заболевания: депрессия (псевдодеменция)*, шизофрения*, конверсионные реакции*
* Дегенеративные заболевания: болезнь Гентингтона, болезнь Пика, прогрессирующий надъядерный паралич, рассеянный склероз, врожденная атаксия (некоторые формы), боковой амиотрофический склероз (некоторые формы), дегенерация базальных ядер.
* Различные заболевания: васкулиты*, острая интермиттирующая порфирия* Примечание: * потенциально обратимая деменция после лечения
основного заболевания
Диагностический алгоритм деменции
Болезнь Альцгеймера (БА)
Синоним — деменция альцгеймеровского типа представляет собой наиболее распространенную форму первичных дегенеративных деменций позднего возраста, которая характеризуется постепенным малозаметным началом в пресенильном или старческом возрасте, неуклонным прогрессированием расстройств памяти и высших корковых функций вплоть до тотального распада интеллекта и психической деятельности в целом, а также типичным набором нейропатологических признаков.
Этиология и патогенез
Несмотря на огромный объем накопленных в последние десятилетия знаний о биологических основах БА, необходимо признать, что этиология подавляющего большинства случаев заболевания остается до сих пор неизвестной. В свете развиваемой в настоящее время концепции клинико-генетической гетерогенности болезни вполне вероятно, что речь идет об этиологически различных формах деменций альцгеймеровского типа, которые развиваются по общим или даже только по частично совпадающим патогенетическим механизмам, приводят к эквифинальным последствиям в виде общего стереотипа развития болезни, сходства клинической и нейроморфологической феноменологии.
Установлено, что БА включает несколько генетически гетерогенных форм (A. Roses и соавт., 1992; Е.И. Рогаев, 1999). Для семейных форм с ранним началом болезни (условно до 65 лет, но чаще в возрасте 40-55 лет) характерен аутосомно-доминантный характер наследования, при котором причиной развития болезни является мутация в единственном гене. Указанные формы составляют лишь небольшую часть (до 10%) патологии, объединяемой в настоящее время под рубрикой БА. При более редких семейных формах с поздним (после 65 лет) началом заболевания тип наследования определяется как олигогенный (с главной мутацией в одном или нескольких генах и модификационным эффектом в других). По мнению специалистов (Е.И. Рогаев, 1999), так называемые спорадические случаи, к которым относится подавляющее большинство пациентов с БА, также могут быть обусловлены мутациями или полиморфизмами в генах, однако патогенная экспрессия генетической аномалии у них находится под влиянием других генов и/или средовых факторов. Недавние исследования в области молекулярной генетики БА привели к идентификации трех генов, ответственных за развитие семейных (т.е. наследственно-обусловленных) форм заболевания. На 21-й хромосоме локализован ген амилоидного предшественника (0-АРР); на 14-й — ген пресенилин-1 (PSN-1) и на 1-й хромосоме — пресенилин-2 (PSN-1).
Носители мутаций в гене АРР встречаются в 3-5% всех семей с пресенильным типом заболевания. Наследование в этих семьях происходит по аутосомно-доминантному типу. Мутации в гене PSN-1 оказались ответственны за 60-70% всех ранних (пресенильных) случаев семейной формы БА (Н.Н. Рязанская и соавт., 1999). Установлено, что мутации в гене PSN-2 более редки. Они обнаружены к настоящему времени только в итальянских семьях и в семьях поволжских немцев. Мутации в гене PSN-1 характеризуются полной пенетрантностью и обязательно проявляются в возрасте от 30 до 50 лет. Мутации в гене PSN-2 характеризуются неполной пенетрантностью, они вовлечены в развитие более редких как ранних, так и поздних семейных форм БА.
Роль мутаций или полиморфизмов в пресенилинах в развитии спорадических случаев поздней БА (т.е. сенильной деменции альцгеймеровского типа) пока остается невыясненной.
Функции генных мутаций и их роль в пусковых механизмах болезни еще недостаточно ясны. Обнаружено, что некоторые мутации в гене белка-предшественника р-амилоида (р-АРР) ответственны за увеличение продукции р-амилоида, из которого формируются так называемые сенильные или амилоидные бляшки, представляющие собой один из двух главных нейроморфологических феноменов заболевания. Полагают, что отложения р-амилоида в виде агрегированных скоплений (сенильных бляшек) в экстрацеллюлярных пространствах коры головного мозга обладают нейротоксичностью и ответственны за развитие дегенеративных изменений в близлежащих нейронах.
Сам по себе р-амилоид представляет собой продукт физиологического протеолитического разрушения высокомолекулярного белка р-АРР. Однако лишь вызванная мутациями в гене р-АРР гиперпродукция р-амилоида или удлинение его молекулы за счет присоединения двух дополнительных аминокислот приводит к патологическому процессу усиленного образования амилоидных бляшек, поскольку удлиненные пептиды агрегируют значительно чаще, чем более короткие их формы.
Идентифицированный недавно е4-изоморфный вариант гена аполипопротеина Е (АроЕ) признан в настоящее время главным генетическим фактором риска подверженности поздней БА.
АроЕ — белок с множественными функциями, который экспрессируется в головном мозге, но не в нейронах, а в глиальных клетках. АроЕ участвует в процессах регенерации при повреждениях центральной нервной системы. Получены доказательства участия АроЕ в компенсаторном холинергическом синаптогенезе (J. Porier и соавт., 1993). Показана взаимосвязь генотипа АроЕ и холинергического дефицита при БА; снижение активности ацетилхолинтрансферазы в гиппокампе и височной коре обратно пропорционально числу копий аллеля е4 гена АроЕ (J. Porier и соавт., 1998). Непосредственные молекулярные механизмы, взаимодействующие с продуктами пресенилинов, АРР или АроЕ, еще ждут исследования на адекватных клеточных моделях или моделях трансгенных животных. Тем не менее, несомненно, что все открытые генетические аномалии так или иначе влияют на процессы, связанные с нарушениями в амилоидных превращениях, которые приводят к образованию нейротоксических амилоидных бляшек.
Признание найденный генетических мутаций этиологическими факторами, по крайней мере, части случаев БА, основано на предположении о том, что аномальный процесс амилоидогенеза является ключевым патогенетическим звеном заболевания. В соответствии с этой гипотезой аномальный амилоидогенез предшествует нейрофибриллярным изменениям, выступая в качестве причины нейрональной дисфункции и последующей гибели нейронов.
Однако морфометрическое изучение биопсийного и аутопсийного материала показало, что тяжесть деменции альцгеймеровского типа, отражающая прогрессирование заболевания, в большей мере коррелирует не с количеством сенильных (амилоидных) бляшек, а с плотностью нейрофибриллярных клубков и утратой синапсов (E. Masliah и соавт., 1994; E. Masliah, 1995).
По мнению H. Braak и E. Braak (1996), возможно, патогенетически более значимым процессом, вызывающим гибель нейронов и развитие деменции, является не аномальный амилоидогенез, а накопление гиперфосфорилированного нерастворимого таупротеина, который составляет основу парноскрученных филамент, образующих нейрофибриллярные клубки. Доказательством справедливости этой гипотезы служат данные об иерархическом распространении нейрофибриллярной патологии, соответствующей последовательным переходам в развитии болезни от инициальных доклинических симптомов к мягкой и далее к умеренной и тяжелой деменции (H. Braak, E. Braak, 1991, 1996; Вег и соавт., 1993).
Другим нейроморфологическим феноменом, который обнаруживает параллелизм с прогрессированием когнитивного снижения, является уменьшение числа синапсов в лобной и височной коре и в гиппокампе (R. Ierry, 1994). Было изучено, каким образом утрата синапсов в различных морфофункциональных структурах мозга коррелирует с клиническими проявлениями заболевания. На основании результатов такого анализа высказано предположение, что развитие деменции при БА прямо связано с утратой синаптических контактов в специфических корковых и подкорковых областях мозга (E. Masliah, R. Terry, 1993).
Выполненные к настоящему времени многочисленные нейрогистологические и нейрохимические исследования аутопсийного мозга больных с деменцией альцгеймеровского типа позволили установить несколько каскадов биологических событий, происходящих на клеточном уровне, которые предположительно вовлечены в патогенез заболевания: нарушение процессов фосфорилирования белков, изменения в метаболизме глюкозы и активация процессов перекисного окисления липидов. Высказано предположение, что каждый из таких каскадов патологических событий или их совокупность могут в конечном итоге приводить к описанным выше структурным изменениям, которые лежат в основе нейрональной дегенерации и сопровождаются развитием деменции. Предположение о возможной каузальной роли самого фактора старения в развитии первичного нейродегенеративного процесса, приводящего к нейрональной гибели, а на клиническом уровне к развитию деменции, хорошо согласуется с установленными в эпидемиологических исследованиях данными об экспоненциальной зависимости частоты сенильной деменции альцгеймеровского типа от возраста. Помимо связанных со старением нарушений церебрального метаболизма глюкозы, на фоне старения происходит усиление свободнорадикальных процессов, что вносит свой вклад в цепочку патологических событий, характерных для нейрональной патологии при БА.
Идентифицированный недавно е4-изоморфный вариант гена аполипопротеина Е (АроЕ) признан в настоящее время главным генетическим фактором риска подверженности поздней БА.
Показано, что при старении ослабляется контроль над свободнорадикальными процессами, в частности из-за недостаточности а-токоферола или экзогенного повреждения природных антиоксидантных систем в организме. Результатом этих изменений является активация процессов перекисного окисления липидов, что способствует накоплению в организме свободных радикалов — молекул, которые в свою очередь могут вызывать необратимые повреждения как на уровне клетки, так и в организме в целом. В частности, активация процессов перекисного окисления липидов приводит к изменению структурной организации мембран (фосфолипидного состава, микровязкости и ионной проницаемости), нарушению функций мембраносвязанных ферментов и рецепторов, повреждению митохондриальных белков и вследствие этого к клеточному энергетическому дефициту.
При нормальном старении все эти параметры нередко ухудшаются по мере увеличения возраста, но в неблагоприятных условиях, например при стрессе или церебральной ишемии, темп возрастного снижения интенсивности метаболизма глюкозы и нарушения энергетического обмена в мозге резко увеличивается.
В этом смысле само по себе старение может выступать не только в роли фактора риска, а, возможно, даже и в роли единственного этиопатогенетического механизма в развитии большинства поздних форм БА, т.е. сенильной деменции альцгеймеровского типа.