Лечение
направлено на поддержание физической активности пациента и улучшения качества его жизни.
ГКС-преднизолон по 0,75 мг/кг/сут. увеличивает мышечную массу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование болезни.(по схеме через день)
препараты, улучшающие обмен веществ ( вит группы В,Е, аминокислоты, препараты кальция)
применяется лечение прозерином, галантамином(ингибиторы холинэстеразы)
ЛФК. (особенно замедляющая образование контрактур), так же, как и пассивное растяжение больных мышц, массаж, электрофорез прозерина, лидазы, кальция хлорида, ванны, индуктотермия.
+ наблюдение кардиолога обязательно!!!
При наличии контрактур и фиксации суставов показано ортопедическое вмешательство
Миопатия Эрба-Рота
Прогрессирующая мышечная дистрофия Эрба-Рота — аутосомно-рецессивная наследственная миодистрофия, отличающаяся полиморфизмом клинических проявлений и вариативностью скорости прогрессирования симптомов. Может носить нисходящий характер, т. е. начинаться со слабости в проксимальных отделах рук, но чаще имеет стандартный восходящий тип распространения мышечных изменений.
дебют в детском или юношеском возрасте чаще в 14-16 лет
конечностно-поясная миодистрофия прежде всего атрофии мышц тазового пояса
ранний признак утиная походка и др миопатические феномены
в дальнейшем атрофии мышц плечевого пояса, рук (форма
ЛЕЙДЕНА-МЕБИУСА)
Этиопатогенез
до конца не изучен? Субстратом дистрофии Эрба-Рота являются патологические метаболические и структурные изменения в мышечной ткани (миопатия). Они возникают в результате генетических мутаций, приводящих к недостатку или полному прекращению синтеза белков, являющихся необходимыми структурными компонентами миоцитов.(Согласно существующей на сегодняшней день версии, причины дистрофии Эрба-Рота заключаются в генетическом дефекте, передающемся от одного из здоровых родителей – здорового носителя мутировавшего гена в парных неполовых хромосомах или в Х-хромосоме. Это такие гены, как 13q12, 17q12-q21.33, 4q12 и 5q33)
Клиника
Симптомы заболевания вначале неспецифичны и включают в себя общую слабость, слабость мышц спины. Постепенно заболевание прогрессирует. Пациент перестает удерживать спину в нормальном положении, развивается гиперлордоз – переразгибание поясницы кзади.
Достаточно рано изменяется походка. Она становится похожей на «утиную» — переваливание ног из-за слабости мышц бедра и тазового пояса.
Быстро развивается гипотрофия мышц верхнего плечевого пояса. Также постепенно развивается общая гипотрофия, а затем и атрофия мышц. Иногда имеет псевдогипертрофия голеней – замена мышечной массы жировой и соединительной тканью.
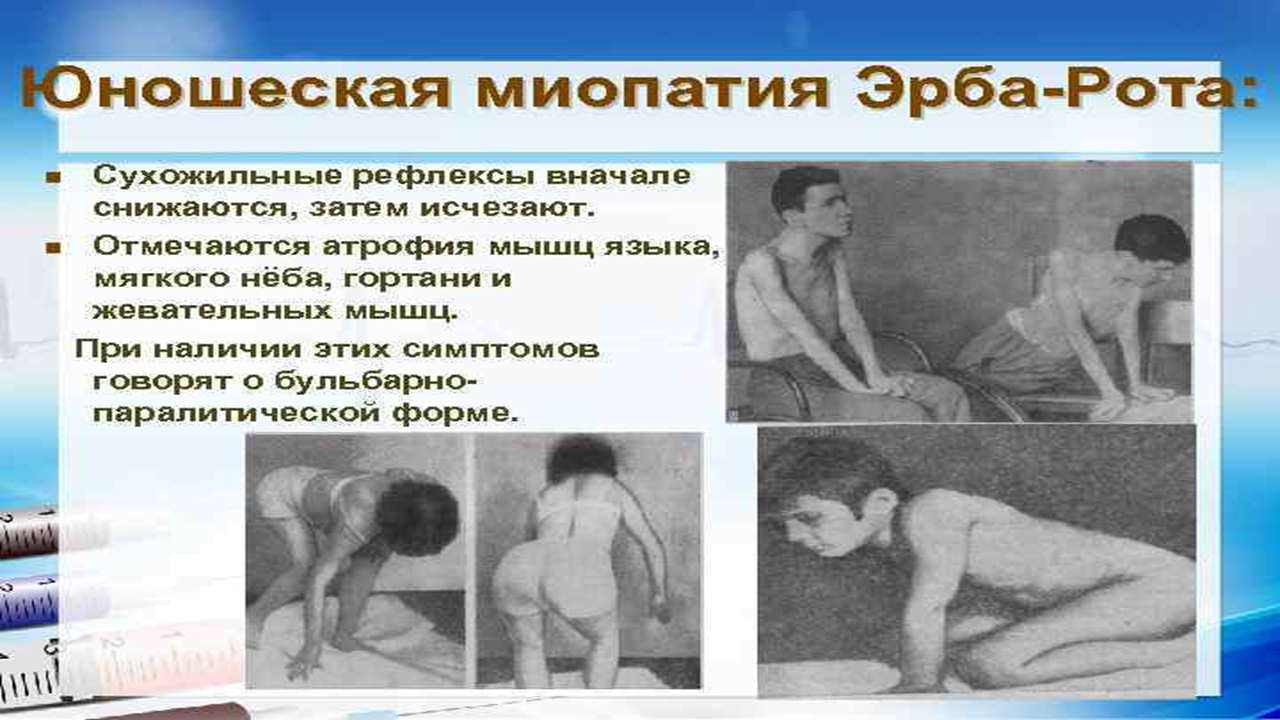
Со временем пациент перестает выполнять многие активные движения. Значимо затрудняется вставание, больным приходится вставать на четвереньки, помогать руками при вставании.
Лицо пациента становится амимично, не полностью смыкаются веки, губы же, напротив, выворачиваются кпереди и нередко утолщаются (губы тапира). Мимику такого пациента ещё иногда называют лицом миопата