

обработка
.docx|
Вопрос 1) Св-ва волновой функ. Понятие об уравнении Шредингера. Квантовые числа как характеристики состояния электрона в атоме. Напишите значения квантовых чисел для каждого из d-электорнов атома … в основном состоянии. Законы движения микрочастиц в квантовой механике выражают волновым уравнением Шредингера. Как и законы Ньютона, это уравнение невозможно вывести из каких-либо более фундаментальных положений. Оно было получено Шредингером на основании анализа аналогии между закономерностями классической механики и оптики.
Уравнение
Шредингера явл. дифференциальным
уравнением в частных производных. Для
стационарного состояния одной частицы
массой m
оно
имеет вид: где: h – постоянная Планка; Ψ – переменная величина; U – потенциальная энергия частицы; Е – полная энергия частицы; x, y, z – координаты. Часто
уравнение Шредингера записывают в
компактной форме:
Переменная Ψ наз. волновой функцией. Её Ψ2 имеет определенный физ. смысл: Ψ2.dv = вероятности рассматриваемой частицы в элементе объёма dv. Величина Ψ2 наз. плотностью вероятности, или электронной вероятности. Ψ должна быть конечной, непрерывной, однозначной, и обращаться в нуль в тех местах пространства, где частица не может находиться. Волновая ф-ия-состояние каждой частицы в микромире, описывается специальной ф-ией(Ψ-функцией) Ψ-амплитуда эл.волны Волновые объекты – размера не имеют и их нельзя охарактеризовать координатами. Волновые объекты или излучения (радиоволны, микроволны, видимое излучение, инфракрасное, у/ф ,рентген., гамма-излучение)-различные проявления одного и того же электромагнитного излучения, отличаются только длиной волны. Св-ва: 1)интерференция-явление наложения волн вплоть до полного погашения; 2)дифракция – способность огибать препятствия, к чему частица вовсе не способна (условие – примерное равенство препятствия и длины волны волнового объекта. Минимальное препятствие для дифракции – атом, т.к. меньше частиц мы не знаем Ψ – функция зависит не только от трех координат, но и от трех целочисленных параметров, названных квантовыми числами. Их обозначение n, l и ml . n – главное квантовое число в значительной степени определяет значение энергии электрона в атоме и размеры электронного облака. Оно принимает значения от +1→+∞. С ростом n возрастают размеры электронного облака и энергия электронов. Кроме цифрового обозначения n используется и буквенная индексация: n=1↔K, n=2↔L, n=3↔M, n=4↔N, …. l – орбитальное (побочное) кв. число, характеризует энергию электрона внутри слоя и определяет форму электронного облака. Оно принимает значения от 0→+(n−1). На практике l чаще обозначается буквами: l=0↔s, l=1↔p, l=2↔d, l=3↔f, l=4↔g, …, l=n−1. ml − магнитное кв. число, определяет ориентацию орбитали в пространстве. Орбиталь же образуется совокупностью электронов с одним и тем же значением магнитного квантового числа, её традиционно обозначают □. При данном значении l, ml принимает значения: 0, ±1→±l . Общее число значений ml : (2l+1).
У
электрона есть и 4-ая кв. характеристика
– спинное кв. число ms,
отражающее 4-ую координату нашего мира
– время. ms
принимает (в единицах атомного мира)
два значения:
Принцип Паули: в атоме не может быть двух электронов с одинаковым набором 4-х квантовых чисел. Правило Хунда: суммарный спин электронов оболочки должен быть максимальным.
|
Вопрос 2) Св-ва волновой функ. Понятие об уравнении Шредингера. Что такое узловые поверхности? Охарактеризуйте квантовыми числами следующее состояние электронов (в основном состоянии): ….
Узловой
поверхностей
орбитали наз. геометрическое место
точек, где волновая функ.
|
Вопрос 3) Дайте опр. следующим понятиям: электронный слой, электронная оболочка, электронное облако, электронная орбиталь, узловая поверхность. Сколько максимально электронов может быть в …-слое, на … - оболочке? Электронный слой (уровень) – совокупность электронов с одним и тем же значением главного кв. числа n. Максимальное число е в слое n: 2n2. n=1: 1-ый слой или К-слой; n=2: 2-ой слой или L-слой; n =3: 3-ий слой или M-слой; и т.д Электронная оболочка – совокупность эл-нов с одним и тем же значением побочного кв. числа l. Мак. число е в оболочке l: 2(2l+1). (2- число значений спинов). l = 0 s-оболочка; l = 1 p-оболочка; l = 2 d-оболочка; l = 3 f-оболочка; l = 4 g-оболочка.
Электронная
орбиталь
– совокупность эл-нов с одним и тем
же значением магнитного кв. числа ml,
её обозначают □. При данном значении
l,
магнитное кв. число принимает значения:
Узловой
поверхностей
орбитали наз. геометрическое место
течек, где волновая функ.
|
Вопрос 4) Принцип Паули и правило Хунда. Сколько максимально электронов может быть в электронном слое, электронной оболочке, на орбитали ? Какую форму имеют s-, p-, d- орбитали ? Принцип Паули: в атоме не может быть двух электронов с одинаковым набором 4-х квантовых чисел. Правило Хунда: суммарный спин электронов оболочки должен быть макссиммальным. Максимальное число е в оболочке l: 2(2l+1). Максимальное число е в слое n: 2n2. Максимальное число на орбитали: 2е. Формы s-, p-, d- орбиталей:
|
Вопрос 5) Энергия электрона в многоэлектронном атоме. Энергический ряд атомных орбиталей. Напишите электронные формулы атомов …, и иона …. Какие степени окисления может иметь … в соединениях? Энергия электрона в атоме водорода (теория Бора) определяется единственным (главным) квантовым числом, а в многоэлектронном атоме – главным и орбитальным квантовыми числами. В целом энергия электрона возрастает по мере роста суммы названных квантовых чисел при доминирующем значении квантового числа. При одинаковой сумме энергия состояния с меньшим значением гл. кв. числа ниже. В соответствии с этими правилами энергетический ряд атомных орбиталей выглядит следующим образом: 1s<2s<2p<3s<3p<4s<3d<4p<5s<4d<5p<6s<4f<5d<6p<7s<5f<6d<7p и т.д.
|
|
Вопрос 6) Современная формулировка периодического закона. Энергия ионизации и сродство к электрону, закономерности в их изменении по периодам и группам периодической системы. Свойства химических элементов, а также формы и свойства образуемых ими соединений, находятся в периодической зависимости от величины заряда ядер их атомов. Энергия ионизации - представляет собой наименьшую энергию, необходимую для удаления электрона от свободного атома в его низшем энергетическом (основном) состоянии на бесконечность. В целом в периоде в соответствии с тенденцией изменения атомных радиусов и увеличением притяжения электронов к ядру должна возрастать. Однако этот рост неравномерен. Энергия, выделяющаяся при добавлении электрона к нейтральному атому, переходящему при этом в отрицательный однозарядный ион, наз. сродством к электрону. Сродство к двум или более электронам отрицательно. Наиболее часто энергию ионизации и сродство к электрону выражают в электроновольтах (эВ). 1 эВ = 1,60.10-19 Дж = 1,6.10-12 эрг. Факторы, определяющие энергию ионизации (энтальпию ионизации): 1) эффект экранирования заряда ядра глубинными оболочками и слоями – уменьшение энергии ионизации атомов в подгруппе . 2) эффект проникновения внешних электронов к ядру. Согласно квантовой механике вероятность нахождения электрона в других слоях кроме «своего» не равна нулю. Этот эффект ведет к упрочнению связи электрона с ядром, т.е. к росту энергии ионизации, (самые проникающие s-электроны, менее – p и еще менее –d). 3) отталкивание электронов одного слоя, особенно находящихся на одной орбитали – уменьшение энергии ионизации. Закономерности в их изменении по периодам и группам периодической системы:
|
Вопрос 7) Атомные и ионные радиусы, как их определяют? Как изменяются радиусы в радах: … Поскольку квантовая механика запрещает точное определение координат частицы, понятия «радиус атома», «радиус иона» условны. Атомные радиусы подразделяют на радиусы атомов металлов, ковалентные радиусы атомов металлов и радиусы атомов благородных газов. Их определяют как половину расстояния между слоями атомов в кристала соответствующих простых в-в. рентгенографическим или нейтронографичским методами. В общем случае радиус атома зависит не только от природы атомов, но и от характера химической связи между ними, агрегатного состояния, температуры и ряда других факторов. Это обстоятельство лишний раз указывает на относительность понятия «радиус атома». Атомы не являются несжижаемыми, неподвижно застывшими шариками, они всегда принимают участие во вращательном и колебательном движении. Радиусы атомов благородных газов значительно больше радиусов атомов неметаллов соответствующих периодов, поскольку в кристаллах благородных газов межатомное взаимодействие очень слабое. Шкала ионных радиусов, понятно, не может быть основана на тех же принципах, что шкала атомных радиусов (ни одна хар-ка индивидуального иона не может быть объективно определена). Современная шкала ионных радиусов основана на допущении, что границей между ионами является точка минимума электронной плотности на линии, соединяющей центры ионов.
|
Вопрос 8) Атомные и ионные радиусы, как их определяют? Основные закономерности их изменения по периодам и группам периодической системы. Поскольку квантовая механика запрещает точное определение координат частицы, понятия «радиус атома», «радиус иона» условны. Атомные радиусы подразделяют на радиусы атомов металлов, ковалентные радиусы атомов металлов и радиусы атомов благородных газов. Их определяют как половину расстояния между слоями атомов в кристала соответствующих простых в-в. рентгенографическим или нейтронографичским методами. В общем случае радиус атома зависит не только от природы атомов, но и от характера химической связи между ними, агрегатного состояния, температуры и ряда других факторов. Это обстоятельство лишний раз указывает на относительность понятия «радиус атома». Атомы не являются несжижаемыми, неподвижно застывшими шариками, они всегда принимают участие во вращательном и колебательном движении. Радиусы атомов благородных газов значительно больше радиусов атомов неметаллов соответствующих периодов, поскольку в кристаллах благородных газов межатомное взаимодействие очень слабое. Шкала ионных радиусов, понятно, не может быть основана на тех же принципах, что шкала атомных радиусов (ни одна хар-ка индивидуального иона не может быть объективно определена). Современная шкала ионных радиусов основана на допущении, что границей между ионами является точка минимума электронной плотности на линии, соединяющей центры ионов. Периодический закон ведёт к след. закономерностям в изменении атомных и ионных радиусов: 1) в периодах слева направо в целом радиус атома уменьшается, затем в конце резко возрастает у атома благородного газа. 2) в подгруппах сверху вниз происходит рост радиуса атома: более значительный в главных подгруппах и менее значительный – в побочных. 3) радиус катиона меньше радиуса атома и уменьшается с ростом заряда катиона. 4) радиус аниона больше радиуса атома. 5) в периодах радиусы ионов d-элементов одинакового заряда плавно уменьшаются, это так называемое d-сжатие. 6) аналогичное явление отмечается и для f-элементов. 7) Радиусы однотипных ионов (имеющих сходную электронную «макушку») в подгруппах плавно возрастают. 8) Если различные ионы имеют одинаковое число электронов (изоэлектронные), то размер таких ионов будет определяться зарядом ядра иона. Наименьшим будет ион с большим зарядом ядра. Радиус изоэлектронных ионов уменьшается с ростом заряда иона.
|
Вопрос 9) Относительная сила кислот и оснований (схема Косселя) на примерах … . Сила кислородных кислот будет возрастать с увеличением степени окисления атома элемента и уменьшается радиуса его иона. У оснований наоборот. Сила бескислородных кислот возрастает с уменьшением степени окисления атома элемента и увеличением радиуса его иона. Сила бескислородных кислот в растворе будет возрастать в подгруппе, т.к при одинаковой степени окисления атома элемента увеличивается радиус его иона. Более сильным электролитам из двух считается тот, у которого при одинаковой молярной концентрации больше степень диссоциации |
Вопрос 10) Основные положения метода валентных связей при описании химической связи. Валентные возможности атомов …. Основные положения метода валентных связей: 1) Образуют химическую связь два электрона с противоположными спинами; происходит перекрывание Ψ-функций и повышение электр. плотности между ядрами; 2) связь локализована в направлении максимального перекрывания Ψ-функций электронов; 3) чем сильнее перекрывание, тем прочнее связь; 4) валентность атома численно равна количеству неспаренных электронов на внешнем слое в основном состоянии или могут быть в возбужденном состоянии |
|
Вопрос 11) Донорно – акцепторный механизм образования ковалентной связи на примерах молекул … , и ионов …. Образование химической связи возможно и за счет пары электронов, принадлежавших до образования связи одному из атомов. Такую связь называют донорно-акцепторной. Первоначало атом А имеет пару е, а атом В – свободную орбиталь. Атом А отдаёт один е атому В, первый становится катионом с неспаренным е, а второй – анионом с неспареннм е. Далее эти частицы традиционным способом образуют полярную ковалентную связь |
Вопрос 12) Гибридизация атомных орбиталей при описании химической связи. Варианты гибридизации с участием s-, p- и d- орбиталей. Какие из приведенных молекул линейные: … ? Несколько разных, но не сильно отличающихся по энергии орбиталей заменяются таким же числом одинаковых по энергии, а значит и симметрично расположенных в пространстве орбиталей. Как видно, гибридная орбиталь более вытянута в сторону связи, следовательно перекрывание будет более сильным, а связь более прочной по сравнению со связью, образуемой негибридной орбиталью. sp – линейная 1800. sp2 – правильный треугольник 1200. sp3 – тетраэдр 109028’. dsp2 – расположение партнеров по вершинам квадрата относительно центрального атома (характерно для комплексных соединений). dsp3 – прав. тригональная пирамида. d2sp3 – октаэдр. Квантово-химические расчеты показывают, что более двух d-орбиталей использовать в гибридной комбинации невыгодно. Поэтому для описания семи и восьми одинаковых связей (например, в комплексах) привлекают в гибридные комбинации f-орбитали: d2sp3f и d2sp3f2. Гибридизация выгодна при n=2, эффективна в большинстве случаев при n=3, малоэффективна при n=4,5,6,7. В гибридную комбинацию практически всегда включают свободные (неподеленные) электронные пары, имеющиеся у атома, в отношении связей которого применяются гибридные представления. Таким образом, для нахождения типа гибридизации орбиталей атома следует просуммировать число его партнеров по химической связи и число свободных электронных пар (у воды 2+2 и sp3 – уголковая, угол 109028’).
|
Вопрос 13) Гибридизация атомных орбиталей при описании химической связи. Варианты гибридизации с участием s-, p- и d- орбиталей. Какие из преведенных молекул плоские: … ? |
Вопрос 14) Гибридизация атомных орбиталей при описании химической связи. Изобратите схемы перекрывания атомных орбиталей при образования связей в молекулах: …
|
Вопрос 15) Образование кратных связей. δ- и π- связи, их особенности. Изобратите схемы перекрывания атомных орбиталей при образования связей в молекулах: … Основные положения метода валентных связей: 1) связь дают два электрона с противоположными спинами; происходит перекрывание Ψ - функций и повышение электр. плотности между ядрами; 2) связь сосредоточена в направлении максимального перекрывания Ψ - функций электронов; 3) валентность атома численно равна количеству неспаренных электронов на внешнем слое в основном состоянии или кол-ву неспаренных электронов, которые могут быть в возбужденном состоянии; 4) чем сильнее перекрывание, тем прочнее связь. По характеру перекрывания орбиталей обычно выделяют σ - и π - связи. Связи, образованные электронными орбиталями, имеющими максимум зоны перекрывания на линии, соединяющей ядра, называются σ - связями. Связи, образованные электронными орбиталями, дающими максимумы перекрывания по обе стороны от линии, соединяющей ядра, называются π - связями. В подавляющем большинстве случаев π - связи менее прочные, чем σ - связи. В кратной связи только одна связь σ - типа, все остальные (одна или две) относятся к π - связям.
|
|
Вопрос 16) Процедура наложения валентных схем в методе валентных связей для объяснения дробной кратности связи на примерах молекул … и иона … Классический метод ВС не способен объяснить дробный характер связи, а многие частицы характеризуются именно такими связями. Был предложен оригинальный подход. Реальное распределение связей в частице представляется результатом наложения двух схем с различной кратностью связей. Например, полуторная = наложение однократной связи и двойной. В молекуле N2O характеристики связи между атомами азота соответствуют кратности 2,5 ((3+2)/2); а связь между атомами азота и кислорода примерно полуторная ((1+2)/2). Подобного распределения связей можно добиться наложением двух валентных схем: N≡N+-O- ↔ N- =N+=O N≡N→O ↔ N←N=O Что приводит к след. распределению связей: N=N-O. Для нитратного аниона: O--N+ ↔O=N+ ↔O--N+ или O--N ↔O=N ↔O←N В результате приходим к выводу, что все связи одинаковые, анион имеет форму правильного треугольника, кратность связи азот-кислород равна 4/3. HN3 : H-N=N+=N-↔H-N--N+≡N или H-N=N=N↔H-N←N≡N , H-N-N=N
|
Вопрос 17) Модель отталкивания локализованных электронных пар (метод Гиллеспи). Основные положения на примере молекул …. Простым и удобным методом предсказания геометрии молекул является модель отталкивания локализованных электронных пар или метод Гиллеспи, имеющий в своей основе метод ВС. Исходными данными для указанного метода являются число связанных с центральным атомом других атомов, валентные возможности всех связанных атомов, количество электронов на внешнем слое центрального атома. Основные положения: 1) Каждая электронная пара, как образующая связь, так и неподеленная, занимает определенное место в пространстве (локализованная электр. пара). Облако двойной и тройной связи рассматривается как единое. Разумеется, электронные пары (эл. облака) отталкиваются. 2) В зависимости от числа локализованных электронных пар они располагаются в пространстве след образом: 2 – линейная конфигурация 3 – правильный треугольник 4 – тетраэдр 5 – правильная тригональная бипирамида 6 – октаэдр 7 – октаэдр с искажением или правильная пентагональная пирамида. Процедура работы: центральный атом – А (самый многовалентный атом), связанный с ним другой атом – В, неподеленная электронная пара – Е; общее число партнеров центрального тома по хим. связи – n; а число неподеленных эл. пар у него – m. ABnEm. Возможные дополнения: а) облако двойной связи занимает в пространстве большее место, чем облако однократной связи. б) облако тройной связи занимает в пространстве большее место, чем облако двойной связи и тем более, чем облако однократной связи. в) в случае полярной ковалентной связи электронное облако сконцентрировано в большей степени возле более электроотрицательного атома. г) облако неподеленной электронной пары занимает в пространстве большее место, чем облако однократной. д) все электроны, образующ. хим. связи, считаются равноценными независимо от их вида (s,p,d,f). е) атомный состав (ядро, внутр. оболочка) не оказывает влияния на расположение валентных электронов. ж) эл. пары располагаются в пространстве таким образом, чтобы отталкивание между ними было минимальным. з) строение молекулы определяется в пространстве связывающих электронных пар
|
Вопрос 18) Эффективные заряды атомов в молекулах. Дипольный момент связи, дипольный момент молекул. Дипольный момент молекулы и её строение на примерах …. Связь, образованная электронами, принадлежащими обоим атомам, называется ковалентной. Ковалентная неполярная возможна лишь между одинаковыми атомами.
А Смещение электронной плотности при образовании полярной ков. связи можно учесть, приписав атомам эффективные заряды в единицах заряда электрона (δ,+δ, -δ). Это условные величины, т.к. электрон нельзя “разделить” между атомами. Эффективные заряды определяют экспериментально и они позволяют представить полярную ковалентную связь комбинацией “чисто” ковалентной и “чисто” ионной связей. Если связь “чисто” ковалентная, то δ=0, если ионная, то δ = ±1 (однократная), δ=±2 (двухкратная связь) и т.п. А для полярной ковалентной связи – промежуточные значения. пример Н+0,2∙∙∙Cl-0,2 0,8*0+0,2*1 (80% ковалентной связи и 20% ионной). Так даже у CsF δ= ±0,99, значит «чисто» ионных соединений нет! Дипольный момент связи (μ) А∙∙∙В равен произведению эффективного заряда на расстояние между ядрами атомов: μ=δ∙d. Дипольный момент молекулы равен векторной сумме дипольных моментов связей с учетом неподеленных электронных пар. Единица μ – один Дебай (1Д), 1Д=3,3∙10-30Кл*м. Обычно μ трудно предсказать, т.к. δ и d изменяются противоположно. Например: HF; HCl; HBr: падение δ (μ падает); CsF; CsCl; CsBr: рост d (μ растет). Что важно – чем симметричнее молекула, тем меньше ее μ, например симметричные молекулы (CO2; BCl3; CCl4; PCl5; SF6) неполярны и имеют μ=0.
|
Вопрос 19) Основные положения метода молекулярных орбиталей (МО ЛКАО). Объясните парамагнитные св-ва … и найдите кратность связи в … и … Основные положения метода МО: 1) При соединении двух атомов в молекулу возможны два состояния – две МО: с более низкой энергией (Есвяз) и более высокой энергией (Еразр). Переход электрона на первую орбиталь ведёт к образованию связи, а на вторую – не даёт. Если атомная орбиталь “переселяется” в молекулу без изменений энергии, она наз. несвязывающей. 2) Метод МО учитывает вклад в хим. связь отдельных е. 3) В первом приближении один электрон на разрыхляющей орбитали сводит на нет действие одного связывающего электрона. Отсюда кратность связи равна полуразности числа связывающих и разрыхляющих электронов.
|
Вопрос 20) Основные положения метода валентных связей при описании химической связи в комплексных соединениях. Рассмотрите на примерах … и … Согласно представлениям метода ВС хим. связь в компл. соед. носит донор-акцепторный характер. Свободные пары е, которыми обладают лиганды, заполняют пустые орбитали центрального иона. Эти орбитали объединяют в гибридные комбинации в зависимости от координационного числа (к.ч). Компл. соед. с к.ч., равным двум, описываются с позиции sp-гибридизации. Компл. соед. с к.ч., равным трём, обычно описываются исходя из sp2-гибрид. Компл. соед. с к.ч., равным четырём, возможны два варианта. Есв – энергия образования связи; Епары – энергия образования электронной пары из неспаренных е. Если Есв < Епары, то спиновое состояние центрального иона в компл. останется тем же, связь в компл. анионе характеризует sp3-гибрид., компл. ион имеет тетраэдрическое строение и характеризуется высоким спином. Если Есв > Епары, то происходит обр. пары, хим. связь в этом компл. описываем с позиций dsp2-гибрид, квадратное строение и характеризуется низким спином. Компл. соед. с к.ч., равным пяти: dsp3-гибрид. Компл. соед. с к.ч., равным шести: ….
|
|
Вопрос 21) Основные положения теории кристаллического поля при описании химической связи в комплексных соединениях. Рассмотрите на примерах … и … Соединения, образованные из двух или более простых соединений, каждое из которых может существовать независимо, называют комплексными соединениями. Центральный ион (атом) в комплексном соединении наз. комплексообразователем. (ион d– или f–элемента, реже p- или s–элемента). Непосредственно окружающие к/о ионы или молекулы, называемые лигандами, образуют вместе с к/о внутреннюю (координационную) сферу (выделяется [ ]). Ионы (молекулы) за пределами внутренней сферы образуют внешнюю сферу компл. соединения. Общее число лигандов во внутр. сфере называется координационным числом. Поскольку комплексообразователем является в большинстве случаев катион металла, а лигандами – анионы или сильно полярные молекулы, то электростатическое взаимодействие вносит существенный вклад в энергетику комплексообразования. Именно на этом акцентирует внимание теория кристаллического поля (ТКП). Её название отражает тот факт, что электростатическое взаимодействие характерно в первую очередь для кристаллов ионных соединений. Основные положения: 1. связь между к/о и лигандами рассматривается как электростатическая. 2. Лиганды считаются точечными ионами или точечными диполями, их электронное строение игнорируется. 3. Лиганды и к/о считаются жестко закрепленными. 4. Подробно рассматривается электронное строение к/о.
|
Вопрос 22) Эквиваленты в-в в реакциях обмена (или в окислительно – восстановительных реакциях). Фактор эквивалентности, молярная масса эквивалента, молярный объём эквивалента. Приведите три примера. Закон эквивалентов. Эквивалентом наз. реальную или условную частицу, соответствующую одному электрону в данной ОВР, или одному протону (одному гидроксилу, одному единичному заряду) в данной обменной реакции. Иными словами, эквивалент – это часть молекулы, приходящаяся на один электрон в данной ОВР или на один протон (одн гидроксил, единичный заряд) в данной обменной реакции. Фактор эквивалентности показывает, какую часть составляет эквивалент от молекулы. Эта величина меняется от нуля до единицы. 1 моль экв. содержит 6,02.1023 эквивалентов, а его масса в граммах и будет молярной массой эквивалента: Мэкв = fэкв.M. Молярный объём эквивалента – это объём, который занимает 1 моль экв. в-ва при данных условиях. Закон эквивалентов: число эквивалентов участников процесса есть постоянная величина |
Вопрос 23) Закон эквивалентов. Различные формы записи закона (реакции в-в в растворах, реакции в-в в газообразном состоянии). Что такое нормальная концентрация и как она связана и молярной концентрацией ? Закон эквивалентов: число эквивалентов участников процесса есть постоянная величина.
1
моль экв. содержит 6,02.1023
эквивалентов, а его масса в граммах и
будет молярной массой эквивалента:
Мэкв
= fэкв.M.
Число молей эквивалентов каждого из
участников процесса может быть найдено
следующим образом:
Если
участники процесса находятся в
растворе, то число молей эквивалентов
каждого из них может быть найдено
умножением нормальной концентрации
в-ва на объём его раствора. В результате
для этого частного случая закон
эквивалентов принимает форму:
Для
химических расчетов с участием газов
наряду с молярными массами активно
использует величина 22,4 л (объём 1 моль
газа при нормальных условиях). Аналогично
вводится:
Нормальная
концентрация
(нормальность) н раствора равна числу
молей эквивалентов растворенного
в-ва в 1 л раствора. Мэкв
= fэкв.M
|
Вопрос 24) Классификация окислительно – восстановительных реакций. Преведите по 2 примера реакций каждого типа (не используйте уравнения из задания №5). Классификация ОВР: 1) межмолекулярные ОВР – в этих реакциях окислителем и восстановителем являются разные молекулы.
2)
внутримолекулярные ОВР – в этих
реакциях окислителем и восстановителем
являются атомы различных или одиноковых
элементов, находящихся в разных частях
одной молекулы, например: (N-3H4)2Cr+62O7
3)
реакции диспропорционирования, в
которых окислителем и восстановителем
являются одни и те же атомы в молекуле:
3Cl02
+ 6KOH
|
Вопрос 25) Типичные восстановители в ОВР. Каковы продукты их окисления? Приведите примеры. Классификация ОВР. Восстановитель – вещество, молекулы или ионы которого отдают электроны. Типичные восстановители: 1) в-ва, молекулы которых содержат атомы элементов в высоких отрицательных степенях окисления или степени окисления которых легко повышаются, например: Na2S-2; KI-1; N-3H3; KN+3O2; K2S+4O3; 2) катионы металлов более низкой степени окисления, например: Fe+2; Sn+2; 3) металлы, из них в первую очередь – щелочные и щелочно-земельные металлы, а так же – водород при повышенных температурах. Продукты их окисления: Если элемент является окислителем – его степень окисления понижается; если элемент является восстановителем – его степень окисления повышается. Среди простых веществ к типичным восстановителям принадлежат активные металлы (щелочные и щелочноземельные, алюминий, цинк, железо и др.), а также некоторые неметаллы, такие как Н2, С (в виде угля или кокса), Р, Si. При этом в кислой среде металлы окисляются до положительно заряженных ионов. В щелочной среде металлы, которые образуют амфотерные гидроксиды (например, цинк, алюминий, олово), входят в состав анионов гидроксокомплексов. С чаще всего окисляется до монооксида или диоксида; Р, при действии сильных окислителей, окисляется до ортофосфорной кислоты. В бескислородных кислотах (HCl, HBr, HI, H2S) и их солях носителями восстановительной функции являются анионы, которые, окисляясь, обычно образуют простые вещества. В ряду галогенид-ионов восстановительные свойства усиливаются от Cl– к I–. Окислительно-восстановительная двойственность – способность одного и того же вещества, в зависимости от реагентов и от условий проведения реакции, выступать как в роли окислителя, так и в роли восстановителя. В таких веществах содержится элемент в промежуточной степени окисления . Окислительно-восстановительная двойственность характерна для простых веществ – неметаллов. Например, азотная кислота за счет азота в высшей степени окисления +5 может выступать только в роли окислителя. В аммиаке азот в низшей степени окисления –3, и, поэтому, за счет азота аммиак может выступать только в роли восстановителя. А в азотистой кислоте HNO2 азот находится в промежуточной степени окисления +3. Азотистая кислота окисляется кислородом, и в этом случае азот – восстановитель |
|
Вопрос 26) Типичные окислители в ОВР. Каковы продукты их восстановления? Классификация ОВР. Приведите примеры. Окислитель – вещество, молекулы или ионы которого принимают электроны. Типичные окислители: 1) в-ва, молекулы которых содержат атомы элементов в высших положительных степенях окисления, например: KMn+7O4, KBi+5O3, K2Cr2+6O7, Pb+4O2; 2) катионы металлов более высокого заряда (более высокой степени окисления), например: Fe+3; Au+3; Sn+4; 3) галогены и кислород (при повышенных температурах). Классификация ОВР: 1) межмолекулярные ОВР – в этих реакциях окислителем и восстановителем являются разные молекулы.
2)
внутримолекулярные ОВР – в этих
реакциях окислителем восстановителем
являются атомы различных или одиноковых
элементов, находящихся в разных частях
одной молекулы, например: (N-3H4)2Cr+62O7
3)
реакции диспропорционирования, в
которых окислителем и восстановителем
являются одни и те же атомы в молекуле:
3Cl02
+ 6KOH
Продукты их восстановления: Если элемент является окислителем – его степень окисления понижается; если элемент является восстановителем – его степень окисления повышается. Среди простых веществ окислительные свойства характерны для типичных неметаллов (F2, Cl2, Br2, I2, O2, O3). Галогены, выступая в качестве окислителей, приобретают степень окисления –1, причем от фтора к иоду окислительные свойства ослабевают. Кислород, восстанавливаясь, приобретает степень окисления –2 (H2O или OH–). Сложные вещества, используемые в качестве окислителей, очень часто, содержат элементы в высшей степени окисления. Например: KMn+7O4; K2Cr+62O7; HN+5O3; KCl+7O4. Концентрированная серная кислота проявляет окислительные свойства за счет серы в высшей степени окисления +6. Продуктами восстановления серы могут быть: SO2 (степень окисления серы +4), сера – простое вещество (степень окисления серы 0), сероводород (степень окисления серы –2). Азотная кислота проявляет ок ислительные свойства за счет азота в высшей степени окисления +5, причем окислительная способность HNO3 усиливается с ростом ее концентрации. Состав продуктов восстановления азотной кислоты зависит от активности восстановителя, концентрации кислоты и температуры системы; чем активнее восстановитель и ниже концентрация кислоты, тем глубже происходит восстановление азота. Водород в степени окисления +1 выступает как окислитель преимущественно в растворах кислот (как правило, при взаимодействии с металлами, расположенными в ряду напряжений до водорода).
|
Вопрос 27) Общие сведения о комплексных соединениях: комплексообразователь, лиганды, координационное число, внутренняя и внешняя сферы. Классификация комплексных соединений. Приведите примеры. Соединения, образованные из двух или более простых соединений, каждое из которых может существовать независимо, называют комплексными соединениями. Центральный ион (атом) в компл. соединении наз. комплексообразователем. (ион d– или f–элемента, реже p– или s–элемента). Непосредственно окружающие к/о ионы или молекулы, называемые лигандами, образуют вместе с к/о внутреннюю (координационную) сферу (выделяется [ ]). Ионы (молекулы) за пределами внутренней сферы образуют внешнюю сферу компл. соединения. Общее число лигандов во внутр. сфере называется координационным числом. Классификация комплексных соединений. Приведите примеры: см. вопрос 28 |
Вопрос 28) Классификация комплексных соединений: по виду координируемых лигандов, по заряду комплексного иона, по классам соединений. Номенклатура комплексных соединений. Приведите примеры. Классификация комплексных соединений: 1) по виду координируемых лигандов: +) аквакомплексы, в которых лигандом явл. молекула воды. Например: K2[Sn(OH)4], Na3[Cr(OH)6]. +) аммиачные комплексы: лиганды NH3 . Например: [Ag(NH3)2]Cl, [Cu(NH3)4](OH)2 . +) ацидокомплексы, в которых лиганды – анионы кислотных остатков. Например: K[Au(CN)2], K2[HgI4]. 2) по заряду комплексного иона: +) катионные – комплексы имеют катионные к/о. Например: [Co(NH3)6]Cl3 +) анионные – комплексы имеют анионные к/о. Например: K[PF6] +) нейтральные – к/о не имеют заряда. Например: [] +) комплексы без внешней сферы, т.е внутрисферные к.с. Например: [Pt(NH3)2Cl2] 3) по классам соединений: +) кислотные комплексы. Например: H2[SiF6]. +) комплексы основания. Например: [Ag(NH3)2]OH. +) комплексы соли. Например: K2[Fe(CN)6]. Простое объяснение механизма образования хим. связи – донорно-акцепторное. Лиганды поставляют свободные электронные пары на пустые d-орбитали к/о. Число координационных мест называется дентатностью (монодентатные, бидентатные и т.п.) Координационные числа обычно в четные и равны удвоенной степени окисления к/о. По характеру лигандов бывают аквакомплексы, гидроксокомплексы, аммиачные комплексы, ацидокомплексы (лиганды – анионы кислотных остатков). Бывают однородные, а бывают и с разными лигандами. Бывают комплексные соединения и без внешней сферы. Названия комплексов строятся по принципу названий солей с указанием лигандов и степени окисления к/о. При построении названия компл. катиона или аниона первоначально называют ионные лиганды в порядке возрастания степени их сложности. При этом молекулы H2O и NH3 обозначаются “аква” и “аммин”. В конце названия компл. катиона или аниона наз. ион-к/о с указанием его степени окисления римскими цифрами в скобках. В случае аниона берется корень латинского названия элемента, к нему добавляется “ат” (станнат, аурат, плюмбат). Например: К3[Al(OH)6] - гексагидроксоалюминат (III) калия; [Cr(NH3)4Cl2]Cl – хлорид дихлоротетрамминхрома (III).
|
Вопрос 29) Закон Гесса, условия его выполнения. Энтальпии образовании, сгорания, атомизации (определение). Закон Гесса: энтальпия (внутренняя энергия) химической реакции не зависит от пути перехода от начальных веществ к продуктам реакции, а определяется только видом и состоянием этих исходных веществ и продуктов. Закон Гесса справедлив для хим. процессов, происходящих при V = const или при Р = const. Предполагается также, Т = const и что система не совершает никакой работы, кроме работы против внешнего давления, связанной с изменением объёма при Р = const. Энтальпия образования – ΔН реакции образования 1 моль данного в-ва из простых в-в при данной температуре. При этом безразлично – осуществим процесс или нет. Обозначение: ΔНобр. Энтальпия сгорания – это ΔН сгорания 1 моль данного в-ва при данной температуре с образованием СО2(г) и Н2О(ж); состав остальных в-в указывают специально. Энтальпия атомизации или энергия ионизации – это наименьшая энергия, необходимая для удаления электрона от свободного атома в его низшем энергетическом (основном) состоянии на бесконечность |
Вопрос 30) Закон Гесса. Следствия из закона Гесса. При каких условиях выполняется этот закон? Закон Гесса: энтальпия (внутренняя энергия) химической реакции не зависит от пути перехода от начальных веществ к продуктам реакции, а определяется только видом и состоянием этих исходных веществ и продуктов. Закон Гесса справедлив для хим. процессов, происходящих при V = const или при Р = const. Предполагается также, Т = const и что система не совершает никакой работы, кроме работы против внешнего давления, связанной с изменением объёма при Р = const. Следствия из закона Гесса: Следствие 1.
Следствие
2.
Следствие 3. ∆Нпрямого процесса = −∆Нобратного порцесса Стандартная энтальпия химической реакции равна разности стандартных энтальпий образования продуктов реакции и реагентов (с учетом стехиометрических коэффициентов).
|
|
Вопрос 31) Стандартные термодинамические характеристики. Понятие о стандартном состоянии индивидуальных жидких и кристаллических в-в, газов и растворов. Закон Гесса. Раздел химии, занимающийся изучением тепловых эффектов процессов – термохимия. Закон Гесса: энтальпия (внутренняя энергия) химической реакции не зависит от пути перехода от начальных веществ к продуктам реакции, а определяется только видом и состоянием этих исходных веществ и продуктов. Особое значение имеют стандартные величины, т.е. такими, где все участники процесса находятся в стандартных состояниях. Для индивидуальных жидких и кристаллических в-в за стандартное состояние принимают реальное состояние этого в-ва при данной т-ре и давлении 1 атм. Со станд. состоянием газов и жидкостей (р-ры несколько сложнее), эти понятия восходят к понятиям “идеальный газ” и “идеальный р-р”. Любой идеальный газ описывается уравнением pV = vRT. Для каждого реального газа уравнение состояния своё и его крайне сложно установить. Поэтому все проблемы отклонения св-в реальных газов от идеальных “прячут” в эффективную величину f (аналог давления). Этот путь используют для реальных газов fV=vRT. Фугитивностью (летучестью) называют такую величину, которая связана с другими термодинамическими характеристиками реального газа так же, как с ним связано давление в случае идеального газа. За стандартное состояние газообр. в-ва принимают состояние гипотетически идеального газа, летучесть которого равна единице, а энтальпии реального газа при той же температуре и давлении, стремящейся к нулю. Т.е. за станд. сост. принимается бесконечно разряженный газ. Та же самая ситуация и с растворами, только вместо летучести берут активность, которая связана с другими хар-ками реального р-ра так, как связана с ними концентрация в случае идеального ра-ра. Осталась одна проблема – аллотропные модификации. Какую из них брать за стандарт? Берут наиболее устойчивую форму, искл.: фосфор берут белый, а не более устойчивый красный, т.к. он более реактивный; S (к.ромб.), а не S (к. моноклин.); С (к. графит), а не С (к. алмаз). Если все участники процесса наход. в стандарт. состоянии, тогда реакция – стандартная и обозначается верхним правым “ноликом”.
|
Вопрос 32) Энтальпия и энергия Гиббса, их физический смысл, связь между ними.
Сумма
внутренней энергии и произведения
объёма в-ва на внешнее давление наз.
энтальпией: Н
= U
+ pV.
Энтальпия, подобно объему, давлению,
температуре и внутренней энергии,
явл. характеристикой состояния системы.
Абсолютные значения Н
не известны, т.к точно не известна
величина нрутренней энергии U.
Научное и прак. значение имеет изменение
энтальпии в ходе процесса, т.е разность:
Энергия
Гиббса
− это термодинамический потенциал
следующего вида:
При
постоянных температуре и давлении:
TΔS
≥ ΔU
+ РΔV
+ Aпол
Физический смысл энтальпии и энергии Гиббса: физический смысл энтальпии: ее изменение — это тепло, подведенное к системе в изобарическом процессе (при постоянном давлении). Энергия Гиббса в равновесном процессе с точностью до знака равна полезной работе, которую может совершить система |
Вопрос 33) Энергия Гиббса как термодинамическая функция состояния. Определение и свойства. Вычисление стандартной энергии Гиббса процесса по справочным данным. Критерий самопроизвольного протекания реакций.
При
постоянных температуре и давлении:
TΔS
≥ ΔU
+ РΔV
+ Aпол
Св-ва ф-ции: 1) однозначная, конечная, непрерывная ф-ция состояния системы; 2) обладает св-вом независимости ΔG от пути перехода от начальных в-в к продуктам. 3) −Aпол ≥ G2 − G1 = ΔG. Значит ΔG для обратимого процесса равно полезной работе системы. Физический смысл энергии Гиббса вытекает из соотношения (*) – энергия Гиббса в равновесном процессе с точностью до знака равна полезной работе, которую может совершить система. В случае протекания неравновесных процессов энергия Гиббса будет (с обратным знаком) равна максимально возможной полезной работе, которую может совершить система. ΔGхим.реакции = ∑niGi(продуктов) − ∑njGj(исх. в-в) .
|
Вопрос 34) Критерий самопроизвольного протекания реакций, энтальпийный и энтропийный факторы процесса. Какие реакции протекают самопроизвольно в водных растворах? При постоянных температуре и давлении: ΔG ≤ −Апол. . При отсутствии полезной работы получаем: ΔG ≤ 0. Из этого соотношение следует, что при протекании обратимых хим. реакций ΔG = 0, а при протекании реальных, самопроизвольных процессов она уменьшается. Иными словами, при Р = const, Т = const самопроизвольно протекают только процессы, сопровождающиеся ΔG < 0. Пределом протекания процесса будет состояние с минимальным значением G при данных условиях. После достижения минимума G перестает изменяться и ΔG = 0, система находится в состоянии равн.
ΔG
= ΔН – ТΔS.
+) ΔН<0 и ΔS>0 ; +) ΔН>0 и ΔS>0, если ТΔS > ΔН ; +) ΔН<0 и ΔS<0, если |ΔН| > |ТΔS|. Чем отрицательней ΔG, тем проще, в более мягких условиях идет процесс. Энтальпийный и энтропийный факторы процесса: Если ΔН<0 отражает стремление к объединению частиц в более крупные агрегаты, то ΔS>0 отражает стремление к беспорядочному расположению частиц, к их дезагрегации. Переход системы в состояние с минимальной энергией когда ΔS=0, если же ΔН=0, то система самопроизвольно переходит в наиболее неупорядоченное состояние. Каждая их этих противоположных тенденций, количественно выражаемых ΔН и ΔS зависит от природы в-ва и условий протекания процесса (т-ра, давление, соотношение между реагентами и т.д).
Произведение
ТΔS
(кДж/моль) явл. энтропийным фактором
процесса, ΔН – энтальпийным фактором.
В состоянии равновесия: ΔН = ТΔS.
Это уравнение явл. условием равновесия,
характеризует такое состояние данной
системы, когда скорости протекающих
в ней противоположных процессов
становятся равными. Из этого уравнения
Реакции, протекающие самопроизвольно в водных растворах: ?????
|
Вопрос 35) Химическое равновесие. Истинное (устойчивое) и кажущееся (кинетическое) равновесие, их признаки. Приведите примеры. Равновесным называют такое состояние системы, которое не изменяется во времени, и эта неизменность не обусловлена протеканием какого-либо внешнего процесса. Равновесие остается неизменным, пока не изменяются внешние условия. Различают истинное (устойчивое) и кажущееся (кинетическое) равновесие. Истинное равновесие сохраняется неизменным не вследствие отсутствия процессов, а в силу протекания их одновременно в двух противоположных направлениях с одинаковой скоростью. Истинное равновесие имеет следующие признаки: 1. Если нет внешнего воздействия, то система неизменна во времени. 2. Система следует за любыми изменениями во времени внешних условий, сколь угодно малыми они бы ни были, а если воздействие снято, то система возвращается в исходное состояние равновесия. 3. Состояние системы будет одинаковым независимо от того, с какой стороны она подходит к равновесию. Кажущееся равновесие также неизменно во времени при отсутствии внешнего воздействия, однако второй и третий признаки для него не характерны. Примером системы в кажущемся равновесии является пересыщенный раствор: достаточно попадания соринки в такой р-р или встряхивания и начинается выделение из р-ра избыточного растворенного в-ва. При изменении внешних условий равновесие изменяется сообразно новым условиям, или, как говорят, «смещается».
|
|
Вопрос 36) Химическое равновесие. Принцип Ле-Шателье – Брауна и смещение равновесия. Рассмотрите на примере реакции …… Смещение равновесия подчиняется закономерности, называемой принципом Ле-Шателье: “если на систему, находящуюся в устойчивом равновесии, воздействовать извне, изменяя одно из условий, определяющих состояние равновесия, то в системе усилится течение того процесса, который будет ослаблять оказываемое воздействие, а равновесие сместится в соответствующую сторону”. Смещение равновесия: 1) Повышение температуры равновесной системы усиливает течение эндотермического процесса, охлаждение – наоборот. 2) изменение давления существенно сказывается лишь на равновесиях газовых систем. Увеличение давления для них ведет к смещению равновесия в сторону меньшего объёма, падение давления – в сторону большего объёма. 3) увеличение концентрации исх. в-в ведет к смещению равновесия вправо (в сторону продуктов).
|
Вопрос
37)
Константа химического равновесия.
Соотношение величин Кр
и Кс
для газовых равновесий. Связь
Равновесным называют такое состояние системы, которое не изменяется во времени, и эта неизменность не обусловлена протеканием какого-либо внешнего процесса. Равновесие остается неизменным, пока не изменяются внешние условия. За стандартное состояние р-ров (в смысле растворенного в-ва) принят р-р с активностью раств. в-ва равной 1 и св-вами бесконечно разбавленного. Газ – летучесть 1, бесконечно разреженный. Это гипотетические системы. Концентрационная зависимость энергии Гиббса k-компонента газовой смеси или р-ра имеет вид: Gk=gk (T) + RTlnCk (идеальный р-р). Сk→ak (реальный р-р). Gk=gk (T) + RTlnРk (идеальный газ). Рk→fk (реальный газ). Именно ф-ции g(T) неизвестны, поэтому неизвестны абсолютные значения G. А из-за логарифмической зависимости нельзя принять за стандартное состояние нулевые значения. Опираясь на приведенные соотношения, выведем выражение для константы равновесия. Возьмем для примера систему с идеальными газами: ΔGхим.реакции = ∑niGi (продуктов) − ∑njGj (исх.в-в) , где ni, nj – числа молей. (коэфф. в ур.)
При
равн. ΔGх.р
= 0
Аналогично
для константы равн. идеального раствора:
Для
газовых равновесий величины
Кр
и Кс
связаны:
Выражение
энергии Гиббса имеет вид:
или:
При стандартном состоянии
Pi,
Pj,
Ci,
Cj
= 1
|
Вопрос 38) Равновесие диссоциации ассоциированных (слабых) электролитов на примере .... Степень диссоциации, константа диссоциации. Закон разбавления Оствалда. Количественной характеристикой диссоциации является степень диссоциации α, равная отношению числа распавшихся на ионы молекул к общему их числу. Ее обычно выражают либо в долевом исчислении, либо в процентах. К слабым электролитам в водных растворах относят кислоты: угольную, сернистую, сероводородную, серную (по второй ступени), ортофосфорную, все карбоновые к-ты; основания: гидроксиды магния, бериллия, алюминия, аммония, все гидроксиды d-элементов.
Константа
равновесия диссоциации электролита:
KtnAm(k)
Отрыв
протона от анионов происходит в меньшей
степени, чем отрыв от нейтральной
молекулы. Кроме того, ионы водорода,
образовавшиеся в первой ступени
диссоциации, подавляют вторую степень
диссоциации, ионы водорода, образовавшиеся
при первой и второй ступенях, подавляют
третью ступень диссоциации. В результате
реально диссоциация протекает лишь
по первой ступени.
Как вывод,
диссоциацию любого слабого электролита,
независимо от того, диссоциирует он
ступенчато или нет, можно смоделировать
равновесием диссоциации симметричного
электролита: KtA
Пусть
общая концентрация электролита KtA
составляет с моль/л; степень диссоциации
при этой концентрации равна α.
Тогда равновесные концентрации
катиона, аниона и недиссоциированного
электролита составят: [Ktn+]
= [An-]
= αс;
[KtA]
= с – αс.
Подставляем эти соотношения в выражение
для константы диссоциации:
Соотношение, связывающее константу диссоциации с концентрацией электролита и степенью его диссоциации носит название закона разбваления Оствальда. Зачастую степень диссоциации элетролита мала, тогда выражение упрощается Кравн ≈ а2с . (а≪1)
|
Вопрос 39) Принципы построения шкалы стандартных термодинамических функций образования ионов в водных растворах. Как определить стандартную энтальпию образования …. в водном растворе. Бесконечно разбавленный раствор – это такой раствор, в котором 1 моль р-ного в-ва приходится на бесконечно большое число молей р-ля. Его характеристики: 1) все электролиты в нём полностью диссоциированы. 2) взаимодействие между ионами полностью отсутствуют. Любое св-во индивидуального иона, например термодинамическое, не может быть объективно определено. В таких ситуациях прибегают к построению шкалы относительных величин, в которой значение рассматриваемого св-ва для одной из систем постулируется, а значение св-ва других систем отсчитываются от принятого значения. В частности для водных растворов электролитов шкала термодинамических функ. обр. ионов строится на основе следующих допущений:
Аналогичным путем были найдены стандартные энергии Гиббса образования ионов и стандартные энтропии ионов. Хотя энтропий в-в могут быть толко положительными, энтропии ионов могут быть и отрицательными, поскольку они – относительные величины. Константа дисс. любого электролита можеть быть найдена по формуле:
где Кравн. = Кдисс., ∆Gпроцесса как разность стандартных энергий Гиббса обр. ионов и недиссоциированного электролита. Электролит находится в растворе со св-вами бесконечно разбавленного, но при этом не диссоциирует на ионы (раствор, стандартное состояние, гипотетически недиссоциированный). Энтальпия обр. раствора данного соединения определенной концентрации равна стандартной энтальпии обр. этого соединения плюс энтальпия его растворения с обр. раствора нужного состава. Если происходит растворение с образованием бесконечно разбавленного раствора, то получаем стандарную энтальпию обр. соед. в состоянии раствора. Если растворяемое соед. явл. электролитом, то его стандарная энтальпия обр. равна сумме стандартных энтальпий обр. ионов.
|
Вопрос 40) Шкалы величин рН и рОН. Вычисление рН растворов неассоциированных электролитов на примерах ….
Вода
диссоциирует крайне незначительно:
2Н2О
Для
простоты вместо гидроксония используют
негидратированный ион Н+,
т.к. это не влияет на последующие
выводы: Н2О
Кравн. = [Н+][ОН-] = Kw – ионное произведение воды. Замена активностей ионов на концентрации оправдана тем, что вода диссоциирует в очень незначительной степени. Действительно, при 298,15К концентрация ионов Н+ и ОН- в воде составляет 1*10-7моль/л (из 555 миллионов молекул воды диссоциирует лишь одна). ΔG0процесса = ΔG0обрН+ + ΔG0обрОН- − ΔG0обрН2О(ж) = 0 – 157,32 − ( −237,25) = 79,93 кДж
Кw
=
lg[Н+]
+ lg[ОН-]
= −14.
Для любого водного р-ра при 298,15К: рН+ рОН=14. В воде в соответствии с уравнением ее диссоциации соответственно рН = рОH = 7, это нейтральная среда. В кислых растворах [Н+] > 10-7моль/л и рН < 7 (рОН>7). В щелочных растворах [Н+] < 10-7моль/л и рН>7 (рОН<7). Получить растворы с большой концентрацией кислоты не представляет труда, например, Н2SO4 и НNO3 неограниченно растворимы в воде и можно приготовить их водные растворы практически любой молярной концентрации. Однако с ростом концентрации кислоты уменьшается степень ее диссоциации и в реальных системах получить растворы с концентрацией ионов водорода, большей 10 моль/л, практически невозможно. Учитывая это, получаем интервал изменения величин рН: −1 < pH < 15.
|
|
Вопрос 41) Равновесие диссоциации воды. Ионное произведение воды. Шкалы величин рН и рОН.
Вода
диссоциирует крайне незначительно:
2Н2О
Для
простоты вместо гидроксония используют
негидратированный ион Н+,
т.к. это не влияет на последующие
выводы: Н2О
Кравн. = [Н+][ОН-] = Kw – ионное произведение воды. Замена активностей ионов на концентрации оправдана тем, что вода диссоциирует в очень незначительной степени. Действительно, при 298,15К концентрация ионов Н+ и ОН- в воде составляет 1*10-7моль/л (из 555 миллионов молекул воды диссоциирует лишь одна). ΔG0процесса = ΔG0обрН+ + ΔG0обрОН- − ΔG0обрН2О(ж) = 0 – 157,32 − ( −237,25) = 79,93 кДж
Кw
=
lg[Н+]
+ lg[ОН-]
= −14.
Для любого водного р-ра при 298,15К: рН+ рОН=14. В воде в соответствии с уравнением ее диссоциации соответственно рН = рОH = 7, это нейтральная среда. В кислых растворах [Н+] > 10-7моль/л и рН < 7 (рОН>7). В щелочных растворах [Н+] < 10-7моль/л и рН>7 (рОН<7). Получить растворы с большой концентрацией кислоты не представляет труда, например, Н2SO4 и НNO3 неограниченно растворимы в воде и можно приготовить их водные растворы практически любой молярной концентрации. Однако с ростом концентрации кислоты уменьшается степень ее диссоциации и в реальных системах получить растворы с концентрацией ионов водорода, большей 10 моль/л, практически невозможно. Учитывая это, получаем интервал изменения величин рН: −1 < pH < 15.
|
Вопрос 42) Равновесие диссоциации комплексных соединений. Константа устойчивости и константа нестойкости. Реакции образования комплексных соединений. Приведите примеры получения гидроксокомплекса, амминокомплекса и ацидокомплекса. Реакции образования комплексных соединений: комплексные соединения образуются и существуют в растворах при сравнительно большом избытке лиганда. Обычно его берут в несколько раз больше того количества, которое необходимо в соответствии со стехиометрическим соотношением. В результате подавляется диссоциация комплексного соединения, и оно стабилизируется. Разная прочность связи во внутренней и внешней сферах комплексного соединения ведет к различию в характере диссоциации этих частей молекулы. По внешней сфере в водных растворах все комплексные соединения являются сильными электролитами, тогда как диссоциация по внутренней сфере происходит в незначительно степени.
K2[Zn(CN)4]
Константа
равновесия для последнего процесса
(диссоциация комплексного иона)
называется константой нестойкости:
Кравн
= Кнест
=
Константа
равновесия обратного процесса: Zn2+
+ 4CN-
Чем больше Куст. (меньше Кнест.), тем прочнее комплексное соединение, тем слабее оно диссоциирует. Ясно, что произведение Куст. и Кнест. равно единице. Примеры:
|
Вопрос 43) Буферные растворы и их св-ва. Расчет рН буферного раствора состава …. Буферными растворами называют такие электролитные системы, рН которых не меняется при разбавлении и мало меняется при добавлении небольших количеств сильных кислот и оснований. Буферные р-ры на кислую область – смеси растворов слабых кислот и их солей. Буферные растворы на щелочную область – смесь водного раствора аммиака (NH4OH) и солей аммония.
Пусть
у нас р-р слабой одноосновной к-ты и
её соли (НА+NаА).
В р-ре имеются след. равновесия: НА
Наличие в р-ре ионов А- за счет полностью диссоциирующей соли NаА ведет к смещению равновесия диссоциации НА влево (подавлению и без того незначительной диссоциации к-ты НА). Пусть о бщая концентрация к-ты равна Ск-ты; общая конц. соли Ссоли; степень диссоциации к-ты α. Равновесие концентрации равны: [А-] = Ссоли + [Н+] = Ссоли + α.Ск-ты ≈ Ссоли ; так как: α<<1 [НА] = Ск-ты − [Н+] = Ск-ты – α.Ск-ты ≈ Ск-ты .
Константа
дисс. к-ты:
Как видно: 1) при разбавлении р-ра водной ведет к одинаковому уменьшению Ск-ты и Ссоли , а отношение Ск-ты/Ссоли не изменится и рН станет прежним. 2) добавим в буферный р-р несколько капель НСl, при этом часть соли NаА превратится в к-ту НА; в результате Ск-ты немного возрастает, а Ссоли – уменьшится, а отношение Ск-ты/Ссоли и соответственно рН буферного р-ра изменится мало. 3) Подобное произойдет при вливании в буферную смесь нескольких капель р-ра NаОН: Ссоли возрастает, Ск-ты мало уменшится, а отношение Ск-ты/Ссоли и соответственно рН буферного р-ра изменится мало |
Вопрос 44) Равновесие растворения и диссоциации малорастворимого электролита. Произведение растворимости. Связь ПР и растворимости (на примере …..).
KtnAm
(k)
насыщ.р-р насыщ.р-р В водных растворах все малорастворимые электролиты не ассоциированы. Другими словами, средняя стадия написанного равновесия отсутствует.
KtnAm
(k)
Соответствующая константа равновесия с учетом того, что концентрация индивидульаного кристаллического в-ва равна единице, принимает вид: Kравн = [Ktm+]n.[An-]m = произведение растворимости (ПР). Таким образом, произведение растворимости (ПР) есть константа равновесия растворения и диссоциации малорастворимого электролита. Оно численно равно произведению концентраций (активностей) ионов в степенях стехиометрических коэффициентов в насыщенном водном растворе данного малорастворимого электролита. Пусть растворимость электролита равна Р моль/л. Тогда:
[Ktm+]
= nP;
[An]
= mP;
ПР = [Ktm+]n.[An-]m
= (nP)n(mP)m
= nn.mm.Pn+m
.
Отсюда находим связь
ПР с растворимостью:
Если ПК > ПР – выпадет осадок, ПК < ПР − осадок растворится, ПК = ПР − установится равновесие. (ПК = [Ktm+]n.[An-]m. ПК – произведение концентраций).
|
Вопрос 45) Условия выпадения осадка и растворения малорастворимых электролитов. Связь ПР с растворимостью на примере ….
|
|
Вопрос 46) Произведение растворимости как константа равновесия растворения и диссоциации малорастворимого соединения. Связь ПР с растворимостью на примере ….
|
Вопрос 47) Польный (необратимый) гидролиз. Взаимное усиление гидролиза (совместный гидролиз). Приведите примеры. Поляризационное взаимодействие катионов и анионов с сильно полярными молекулями воды ведет к реакции, наз. гидролизом солей. При смешении растворов двух солей, одна из которых – гидролизована по катиону, а другая – по аниону, равновесия гидролиза в этих растворах:
Ktn+
+ H2O
Am-
+ H2O
Как видно, гидролиз первой соли будет усиливать гидролиз второй соли и наоборот. В этом случае говорят о взаимном усилении гидролиза. В этой ситуации образование продукта обменной не возможно, должны образовываться продукты гидролиза. Состав их зависит от большого числа фактов: концентраций сливаемых растворов, порядка смешения, степени перемешивания и т.д.
Примеры:
2FeCl3
+ 3Na2CO3
+ 3H2O
→ 2Fe(OH)3
2CuCl2
+ 2Na2CO3
+ H2O
→ (CuOH)2CO3
|
Вопрос 48) Польный (необратимый) гидролиз. Приведите два примера. Совместный гидролиз двух солей с образованием а) гидроксида металла (+3), б) основного карбоната металла (+2).
|
Вопрос 49) Гидролиз солей одновременно по катиону и аниону (обратимый гидролиз). Расчет константы гидролиза, степени гидролиза и рН растворов таких солей на примере … Поляризационное взаимодействие катионов и анионов с сильно полярными молекулями воды ведет к реакции, наз. гидролизом солей. Гидролиз соли по катиону и по аниону одновременно можно представить в общем виде следующим образом:
Kt+
+ A-
+ H2O
Константа гидролиза имеет вид:
Пусть общая концентрация соли, гидролизованной одновременно по катиону и аниону, равна с моль/л, степень гидролиза составляет h. Тогда:
рН
растворов:
|
Вопрос 50) Гидролиз солей по катиону. Способы подавления гидролиза. Расчет константы гидролиза, степени гидролиза и рН растворов солей, гидролизованных по катиону на примере …. Поляризационное взаимодействие катионов и анионов с сильно полярными молекулями воды ведет к реакции, наз. гидролизом солей. Молекулярная причина гидролиза – поляризационное взаимодействие ионов с сильно полярной молекулой воды. В результате катионы «вырывают» из молекулы воды ОН‑, а анион – Н+. Другими словами, катионы и анионы вторгаются в равновесие диссоциации воды, нарушают его, в результате в случае катиона р-р становится кислым (гидролиз по катиону), в случае аниона – щелочным (гидролиз по аниону). Сила поляризационного вз-ия увеличивается с ростом заряда иона и уменьшением радиуса его иона. Как раз многозарядные катионы (+2,+3) являются катионами слабых оснований, аналогично компактные анионы с зарядом (-2,-3) явл. анионами слабых кислот.
Продукты
гидролиза подавляют вторую ступень,
поэтому гидролиз реально протекает
лишь по первой ступени: Ktn+
+ H2O
Константа
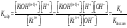
гидролиза − константа равновесия
процесса гидролиза. Пусть с
– общая концентрация катиона (общ.
концентрация соли в случае симметричного
электролита); h
– степень гидролиза катиона Kt.
Равновесные концентрации составляют:
Способы подавления гидролиза: 1) охлаждение раствора; 2) добавление кислоты в раствор для подавления гидролиза по катиону, добавление щелочи в раствор для подавления гидролиза по аниону.
|
|
Вопрос 51) Гидролиз солей по аниону. Способы подавления гидролиза. Расчет константы гидролиза, степени гидролиза и рН растворов солей, гидролизованных по аниону на примере …. Поляризационное взаимодействие катионов и анионов с сильно полярными молекулями воды ведет к реакции, наз. гидролизом солей. Молекулярная причина гидролиза – поляризационное взаимодействие ионов с сильно полярной молекулой воды. В результате катионы «вырывают» из молекулы воды ОН-, а анион – Н+. Другими словами, катионы и анионы вторгаются в равновесие диссоциации воды, нарушают его, в результате в случае катиона р-р становится кислым (гидролиз по катиону), в случае аниона – щелочным (гидролиз по аниону). Сила поляризационного вз-ия увеличивается с ростом заряда иона и уменьшением радиуса его иона. Как раз многозарядные катионы (+2,+3) являются катионами слабых оснований, аналогично компактные анионы с зарядом (-2,-3) явл. анионами слабых кислот.
Продукты
гидролиза подавляют вторую ступень,
поэтому гидролиз реально протекает
лишь по первой ступени: Am-
+ H2O
Константа
гидролиза − константа равновесия
процесса гидролиза. Пусть с
– общая концентрация катиона (общ.
концентрация соли в случае симметричного
электролита); h
– степень гидролиза аниона Аn.
Равновесные концентрации составляют:
Способы усиления гидролиза солей: 1) разбавление раствора соли; 2) нагрев раствора, поскольку энтальпии гидролиза положительны; 3) добавление щелочи в раствор для усиления гидролиза по катиону, добавление кислоты в раствор для усиления гидролиза по аниону |
||||
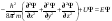 ,
,
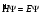 ,
где
,
где
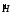 - оператор Гамильтона (гамильтониан),
обозначает все те математические
действия, которые производят в левой
части над величиной Ψ.
- оператор Гамильтона (гамильтониан),
обозначает все те математические
действия, которые производят в левой
части над величиной Ψ.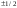 .
Договоримся считать
.
Договоримся считать
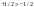 и будем обозначать электроны с
и будем обозначать электроны с
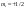 ↑,
↑,
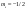 ↓.
↓. (
(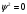 ).
Узловые поверхности могут быть
сферическими, плоскими и коническими.
По определению каждая орбиталь имеет
сферическую узловую поверхность с
бесконечным радиусом. Главное кв.
число равно общему числу узловых
поверхностей данной орбитали.
).
Узловые поверхности могут быть
сферическими, плоскими и коническими.
По определению каждая орбиталь имеет
сферическую узловую поверхность с
бесконечным радиусом. Главное кв.
число равно общему числу узловых
поверхностей данной орбитали.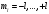 .
Общее число значений составляет
(2l+1).
.
Общее число значений составляет
(2l+1).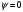 (
(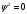 ).
Узловые поверхности могут быть
сферическими, плоскими и коническими.
По определению каждая орбиталь имеет
сферическую узловую поверхность с
бесконечным радиусом. Главное кв.
число равно общему числу узловых
поверхностей данной орбитали.
).
Узловые поверхности могут быть
сферическими, плоскими и коническими.
По определению каждая орбиталь имеет
сферическую узловую поверхность с
бесконечным радиусом. Главное кв.
число равно общему числу узловых
поверхностей данной орбитали.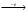 В
: А+
↔ В-
(электростатическое притяжение). Это
ионная связь. Строго говоря, соединения
с чисто ионной связью нет. Самый частый
тип связи – полярная ковалентная. В
этом случае общая электронная пара
смещена к одному из атомов в соответствии
с их электроотрицательностью. ЭО –
это суммарная хар-ка способности атома
отдавать и присоединять электроны.
ЭО сами по себе не очень важны, важны
ΔЭО.
Чем больше ΔЭО,
тем полярнее (ионнее) связь.
В
: А+
↔ В-
(электростатическое притяжение). Это
ионная связь. Строго говоря, соединения
с чисто ионной связью нет. Самый частый
тип связи – полярная ковалентная. В
этом случае общая электронная пара
смещена к одному из атомов в соответствии
с их электроотрицательностью. ЭО –
это суммарная хар-ка способности атома
отдавать и присоединять электроны.
ЭО сами по себе не очень важны, важны
ΔЭО.
Чем больше ΔЭО,
тем полярнее (ионнее) связь.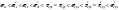
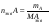 ;
;
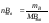 , где mA
и mB
– массы в-в А и B.
И поэтому другая запись закона
эквивалентов имеет: “число
молей эквивалентов участников данного
процесса есть постоянная величина”:
nэкв.A
= nэкв.B
= nэкв.C
= … = const.
, где mA
и mB
– массы в-в А и B.
И поэтому другая запись закона
эквивалентов имеет: “число
молей эквивалентов участников данного
процесса есть постоянная величина”:
nэкв.A
= nэкв.B
= nэкв.C
= … = const.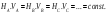
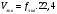 .
.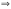
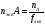
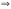
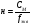 .
.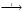 N02
+ Cr+32O3
+
4H20.
N02
+ Cr+32O3
+
4H20.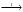 5KCl-1
+ KCl+5O3
+ 3H2O.
5KCl-1
+ KCl+5O3
+ 3H2O.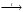 N02
+ Cr+32O3
+
4H20.
N02
+ Cr+32O3
+
4H20.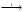 5KCl-1
+ KCl+5O3
+ 3H2O.
5KCl-1
+ KCl+5O3
+ 3H2O. ,
где ni
и nj
– числа молей (коэффициенты в уравнении).
,
где ni
и nj
– числа молей (коэффициенты в уравнении). , где ni
и nj
– числа молей (коэффициенты в уравнении).
, где ni
и nj
– числа молей (коэффициенты в уравнении).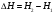 .
.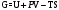 ,
где U
− внутренняя энергия, P −
давление, V −
объем, T −
абсолютная температура, S −
энтропия. Энергию Гиббса можно понимать
как полную химическую энергию системы
(кристалла, жидкости и т. д.)
,
где U
− внутренняя энергия, P −
давление, V −
объем, T −
абсолютная температура, S −
энтропия. Энергию Гиббса можно понимать
как полную химическую энергию системы
(кристалла, жидкости и т. д.)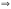 Aпол
≥ ΔU
+ РΔV
– TΔS
= U2
+
РV2
− TS2
−(U1
−
РV1
−
TS1)
(*). Отсюда:
Aпол
≥ ΔU
+ РΔV
– TΔS
= U2
+
РV2
− TS2
−(U1
−
РV1
−
TS1)
(*). Отсюда:
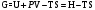 наз. энергией Гиббса. ΔG
≤ −Апол
. Из определении этой функ. следует,
что абсолютное её значение неизвестно,
изменение этой функ. при переходе от
начального состояния к конечному не
зависит от пути перехода.
наз. энергией Гиббса. ΔG
≤ −Апол
. Из определении этой функ. следует,
что абсолютное её значение неизвестно,
изменение этой функ. при переходе от
начального состояния к конечному не
зависит от пути перехода.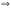 Aпол
≥ ΔU
+ РΔV
– TΔS
= U2
+
РV2
− TS2
−(U1
−
РV1
−
TS1)
(*). Отсюда:
Aпол
≥ ΔU
+ РΔV
– TΔS
= U2
+
РV2
− TS2
−(U1
−
РV1
−
TS1)
(*). Отсюда:
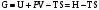 наз. энергией Гиббса. ΔG
≤ −Апол
. Из определении этой функ. следует,
что абсолютное её значение неизвестно,
изменение этой функ. при переходе от
начального состояния к конечному не
зависит от пути перехода.
наз. энергией Гиббса. ΔG
≤ −Апол
. Из определении этой функ. следует,
что абсолютное её значение неизвестно,
изменение этой функ. при переходе от
начального состояния к конечному не
зависит от пути перехода.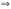 самопроизвольное течение процесса
(ΔG<
0) возможно при:
самопроизвольное течение процесса
(ΔG<
0) возможно при: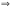
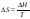 :
расчет изменения энтропии в равновесном
процессе возможен из непосредственно
измеряемых величин. ΔН фазового
перехода можно определить экспериментально
с помощью калориметра.
:
расчет изменения энтропии в равновесном
процессе возможен из непосредственно
измеряемых величин. ΔН фазового
перехода можно определить экспериментально
с помощью калориметра.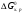 и константы равновесия.
и константы равновесия.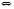 ΔGхим.реакции
=
∑ni[gi
(T)
+ RTlnРi]
− ∑nj[gj
(T)
+ RTlnРj]
= ∑nigi
(T)
− ∑njgj
(T)
+ RT(∑ni.lnРi
−
∑ nj.lnРj)
= Ф(Т) + RT.
ΔGхим.реакции
=
∑ni[gi
(T)
+ RTlnРi]
− ∑nj[gj
(T)
+ RTlnРj]
= ∑nigi
(T)
− ∑njgj
(T)
+ RT(∑ni.lnРi
−
∑ nj.lnРj)
= Ф(Т) + RT.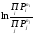 .
.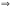
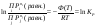
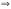
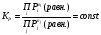 при данной температуре, наз. константой
равновесия. В случае реальных газов
вместо давлений будут фигурировать
фугитивности.
при данной температуре, наз. константой
равновесия. В случае реальных газов
вместо давлений будут фигурировать
фугитивности.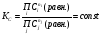 . В случае реальных растворов концентрации
должны быть заменены на активности.
. В случае реальных растворов концентрации
должны быть заменены на активности.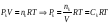
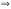
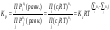 .
.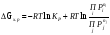 (газы);
(газы);
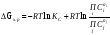 (растворы).
(растворы).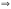
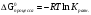
 nKtm+
+ mAn-
называется константой диссоциации:
Кравн
= Кдисс
=
nKtm+
+ mAn-
называется константой диссоциации:
Кравн
= Кдисс
=
 .
.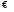 Ktn+
+ An-
.
Ktn+
+ An-
.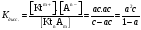 .
.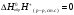 ;
;
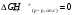 ;
;
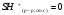

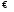 Н3О+
+ ОН-
;
Н3О+
+ ОН-
;
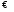 Н+
+ ОН-
.
Н+
+ ОН-
.
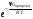 = 1,0*10-14;
[Н+][ОН-]=1,0*10-14
= 1,0*10-14;
[Н+][ОН-]=1,0*10-14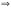 − lg[Н+]
− lg[ОН-]
= 14; рН = −lg[Н+];
рОН = − lg[ОН-].
Отсюда: рН + рОН = 14.
− lg[Н+]
− lg[ОН-]
= 14; рН = −lg[Н+];
рОН = − lg[ОН-].
Отсюда: рН + рОН = 14.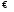 Н3О+
+ ОН-
;
Н3О+
+ ОН-
;
 Н+
+ ОН-
.
Н+
+ ОН-
.
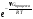 = 1,0*10-14;
[Н+][ОН-]=1,0*10-14
= 1,0*10-14;
[Н+][ОН-]=1,0*10-14 − lg[Н+]
− lg[ОН-]
= 14; рН = −lg[Н+];
рОН = − lg[ОН-].
Отсюда: рН + рОН = 14.
− lg[Н+]
− lg[ОН-]
= 14; рН = −lg[Н+];
рОН = − lg[ОН-].
Отсюда: рН + рОН = 14. 2K+
+ [ Zn(CN)4]2-
; [Zn(CN)4]2-
2K+
+ [ Zn(CN)4]2-
; [Zn(CN)4]2-
 Zn2+
+ 4CN-
.
Zn2+
+ 4CN-
. .
. [Zn(CN)4]2-
наз. константой устойчивости: Кравн.
= Куст.
=
[Zn(CN)4]2-
наз. константой устойчивости: Кравн.
= Куст.
=
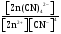 .
. Н+
+
А-
; NаА
Н+
+
А-
; NаА
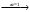 Nа+
+ А-
.
Nа+
+ А-
.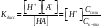 . Отсюда:
. Отсюда:
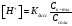 .
. KtnAm
KtnAm
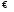 nKtm+
+ mAn-
nKtm+
+ mAn-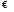 nKtm+
+ mAn-
nKtm+
+ mAn-
 .
.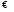 KtOH(n-1)+
+ H+
KtOH(n-1)+
+ H+ HA(m-1)-
+ OH-
HA(m-1)-
+ OH-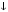 + 6NaCl + 3CO2
+ 6NaCl + 3CO2
 + CO2
+ CO2 + 4NaCl
+ 4NaCl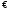 KtOH
+ HA
KtOH
+ HA .
.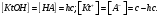 Отсюда:
Отсюда:
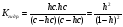 .
Значение константы гидролиза не
зависит от концентраций солей
гидролизованной, или по другому степень
гидролиза соли, подвергающейся
гидролизу по катиону и аниону
одновременно, будет одной и той же при
любых концентрациях соли в растворе.
.
Значение константы гидролиза не
зависит от концентраций солей
гидролизованной, или по другому степень
гидролиза соли, подвергающейся
гидролизу по катиону и аниону
одновременно, будет одной и той же при
любых концентрациях соли в растворе.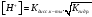
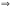
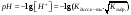 .
.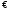 KtOH(n-1)+
+ H+
.
KtOH(n-1)+
+ H+
.
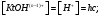
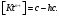 Тогда, получаем:
Тогда, получаем:
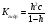 .
.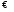 HA(m-1)-
+ OH-
.
HA(m-1)-
+ OH-
.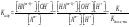
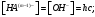
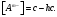 Тогда, получаем:
Тогда, получаем:
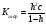 .
.