

5 курс / Госпитальная педиатрия / Атлас_редких_болезней_Педиатрия,_2016_г_
.pdfАтлас редких болезней
Протокол полисомнографии
Орган зрения: помутнение роговицы. У 40% больных выраженное помутнение, что связано с ее утолщением; мегалокорнеа (увеличение роговицы), ретинопатия, изменение диска зрительного нерва, внутриглазная гипертензия, глаукома.
Гепатоспленомегалия. Пациент Т., 11 лет, до получения ферментозаместительной терапии
Центральная нервная система: гидроцефалия, патология спинного мозга.
Сердечно-сосудистая система: прогрессирующая дегенерация клапанов сердца с образованием стеноза. Наиболее часто поражаются митральный и аортальный клапаны. Часто при ЭКГ наблюдается синусовая тахикардия. У 25–35% больных развивается артериальная гипертензия. Кардиомиопатия встречается нечасто.
Желудочно-кишечная система: синдром раздраженного кишечника, увеличение печени и селезенки.
190

Мукополисахаридозы
МРТ головного мозга. |
Т2-взвешенное изображение |
Т1-FLAIR взвешенное |
Т2-взвешенное изображение |
в сагиттальной проекции. |
изображение в коронарной |
в аксиальной проекции. |
Атрофия вещества больших |
проекции. Выраженная |
Выраженное вторичное |
полушарий с расширением |
атрофическая |
атрофическое расширение |
ликворных пространств. |
вентрикуломегалия. Состояние |
боковых желудочков. |
Вторичная демиелинизация |
после ликворошунтирующей |
Формирование субкортикальных |
в субкортикальных отделах |
операции |
кист в лобных долях. Умеренное |
вследствие отложения |
|
расширение периваскулярных |
гликопротеинов. Выраженный |
|
пространств в проекции |
стеноз позвоночного канала |
|
базальных ядер. |
на уровне краниовертебрального |
|
Обращает на себя внимание |
перехода с компрессией |
|
значительное расширение |
спинного мозга |
|
селлярной цистерны |
|
|
Диагностика
•Повышенная экскреция дерматансульфата с мочой.
•В культуре фибробластов или в изолированных лейкоцитах снижение активности N-ацетилгалактозамин-4-cульфатазы (арилсульфатазы В).
•Исследование ДНК.
•Для пренатальной диагностики используют амниоциты и клетки ворсин хориона.
Дифференциальный диагноз:
•другие виды мукополисахаридозов;
•ганглиозидозы.
Лечение
1.В настоящее время внедрены методы ферментозаместительной терапии, которая является наиболее безопасным вариантом. Для ферментозаместительной терапии МПС VI применяется препарат Наглазим (галсульфаза). Галсульфаза — это рекомбинантная форма N-ацетилгалактозамина-4-сульфатазы человека. Препарат вводится в дозе 1 мг/кг один раз в неделю в виде внутривенной инфузии в течение 4 часов в условиях стационара: примерно 2,5% раствора вводят в течение первого часа, остальной объем (примерно 97,5%) в течение последующих 3 часов. Ферментозаместительная терапия требует пожизненного еженедельного выполнения внутривенной инфузии.
191

Атлас редких болезней
2.При необходимости проводится хирургическая коррекция глаукомы, скелетных аномалий, карпального туннельного синдрома, грыжесечение.
3.Коррекция сердечной недостаточности, дыхательных нарушений.
Прогноз. Продолжительность жизни примерно 30 лет.
Список рекомендованной литературы
1.Коннет Л. Джонс. Наследственные синдромы по Дэвиду Смиту. М.: Практика. 2011.
2.Azevedo AC., Kalakun L. et al. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet. 2004; 66: 208–13.
3.Valayannopoulos V., Nicely H., Harmatz P. et al. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010; 5: 5.
4.Braunlin E.A., Harmatz P.R. Cardiac disease in patients with mucopolysaccharidosis presentation, diagnosis and management. J. of Inherited Metabolic Disease. (Jul 2011).
! Для всех форм мукополисахаридоза
1.Необходимо крайне осторожно использовать производные бензодиазепина: серьезным негативным эффектом этих препаратов является угнетение дыхания. У пациентов часто наблюдаются приступы апноэ во сне, препараты усугубляют дыхательные нарушения.
2.Ограничить назначение препаратов, усиливающих метаболизм, так как при этом увеличивается уровень гликозаминогликанов в крови.
3.При остеопорозе препараты кальция назначают в среднетерапевтических, а не высоких дозах!!!
4.Нельзя назначать гормональные препараты, стимулирующие рост — это вызывает усугубление метаболизма гликозаминогликанов и способствует переломам.
192

НАСЛЕДСТВЕННАЯ ТИРОЗИНЕМИЯ I ТИПА
(TYROSINEMIA TYPE I)
МКБ-10: E70.2; OMIM 276700
Определение. Наследственная тирозинемия I типа (НТ1) — нарушение обмена веществ, проявляющееся дефицитом ферментов фумарилацетоацетазы и фумарилацетоацетат гидролазы, гепаторенальной тирозинемией.
Синонимы: отсутствуют.
Эпидемиология. Частота НТ1 в различных популяциях колеблется от 1:100– 200 тысяч живых новорожденных. Частота носительства мутаций НТ1 в популяциях 1:100–150 человек. Половых различий ни в частоте встречаемости, ни в тяжести течения НТ1 не выявлено.
Четких взаимосвязей между генотипом и фенотипом не установлено, различные клинические варианты могут присутствовать у членов одной семьи с одинаковыми мутациями.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Болезнь обусловлена мутациями в гене фермента фумарилацетоацетазы (фумарилацетогидролазы, FAH). Ген FAH, локализованный на длинном плече 15-й хромосомы (15q23–q25), состоит из 14 экзонов. В США 4 частые мутации встречаются у 60% детей с НТ1 типа: c.1062+5G>A (IVS12+5G>A); с.554-1G>T (IVS6-1G>T); c.607-6T>G (IVS7-6T>G); p.Pro261Leu.
Патогенез НТ1 типа заключается в интоксикации продуктами аномального эндогенного распада тирозина. Накопление этих метаболитов приводит к развитию печеночной недостаточности, циррозу, тубулопатии с формированием синдрома Фанкони, полинейропатии, кардиомиопатии, а на более поздней стадии — гепатоцеллюлярной карциноме.
Естественное течение НТ1
Большинство (до 90%) пациентов без специфического лечения и транс-
плантации печени погибали в возрасте до 10 лет. Согласно исследованию |
||
F. J. van Spronsen с соавт. (1994), |
|
|
обследование 108 детей из 125 цен- |
|
|
тров Европы, США, Канады и Японии |
|
|
показало, что выживаемость зависела |
|
|
от сроков появления симптомов. Так, |
|
|
при дебюте до 2-месячного возраста |
|
|
к 1 году умерли практически все дети |
|
|
(96%), при начале заболевания в воз- |
|
|
расте от 2 до 6 мес — выживаемость |
|
|
была чуть выше (до 6 лет умирали |
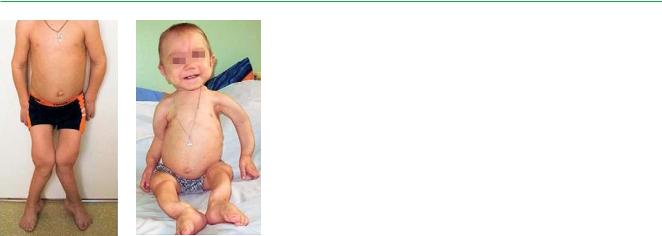
Пациентка К., 8 мес. Мышечная гипотония, |
|
74%), при появлении симптомов в воз- |
||
проявления динамической непроходимости, |
||
расте старше 6 мес в течение 10 лет |
||
гипотрофия, метеоризм и признаки «цветущего» |
||
умирали 38%. |
рахита, гипергидроз |
|
193
Атлас редких болезней
Пациент Б., |
Пациент М., 5 лет |
12 лет. Сочетание |
(вес 8 кг), с НТ1Б. |
клинических |
До начала терапии |
проявлений |
у ребенка |
фосфат-диабета с |
отмечалась |
циррозом печени, |
полиорганная |
повышением |
недостаточность, |
уровня сукцини- |
легочно-сердечная |
лацетона в моче |
недостаточность |
(33 ммоль/моль |
IIБ, надпочеч- |
креатинина при |
никовая недо- |
норме 0–2), |
статочность |
повышением |
(гипертрихоз, |
тирозина в сыво- |
гиперпигментация, |
ротке крови до |
артериальная |
254 Ед (норма |
гипертензия), |
19–200); в гене |
почечный |
FAH мутация IVS |
канальцевый |
6-1G/T в гомо- |
ацидоз в составе |
зиготном состо- |
синдрома Фанкони, |
янии. Диагноз: |
периферическая |
«НТ1Б типа, |
полинейропатия |
осложненная син- |
|
дромом Фанкони» |
|
(до лечения) |
|
Острая форма НТ1А типа:
•начало болезни: первые недели/месяцы жизни;
•задержка развития;
•фебрильная лихорадка;
•рвота, дегидратация;
•диарея или динамическая непроходимость;
•гепатомегалия;
•увеличение размеров живота и его напряжение (как за счет гепатомегалии, в меньшей степени спленомегалии, так и за счет асцита и динамической непроходимости);
•желтуха (на стадии острого гепатита);
•мелена, рвота кофейной гущей (при остром желудочно-кишечном кровотечении);
•безбелковые отеки, анасарка, кровотечения (при снижении белково-синтетической функции печени);
•специфический «капустный» запах тела при высоком уровне метионина;
•смерть в возрасте до 1 года (6–8 мес) от печеночной недостаточности или кровотечения.
Хроническое течение НТ1Б типа:
•чаще с преимущественным поражением печени (циррозом, гепатоцеллюлярной карциномой);
•реже с ведущим поражением почек (рахитоподобного заболевания вследствие тяжелой тубулопатии; вторичного синдрома Фанкони — аминоацидурия, глюкозурия, фосфатурия, почечный канальцевый ацидоз);
•редко заболевание может начаться с непеченочных проявлений по типу порфирии (периферической нейропатии, острых абдоминальных кризов);
•тирозинемические кризы с лихорадкой, рвотой, интоксикацией, кардиомиопатией (нередко провоцируются белковой пищей).
Клинические варианты течения положены в основу классификации НТ1. НТ1 тип А характеризуется острым течением, ранним дебютом (от 2 до 5–7 мес), встречается в три раза чаще, чем тип Б с подострым или хроническим течением. НТ1А типа часто остается нераспознанной или подтверждается после смерти ребенка. Причиной смерти при естественном течении НТ1А типа чаще является острая печеночная недостаточность и катастрофическое кровотечение на ее фоне; диагноз затрудняется такими сопутствующими состояниями, как внутриутробная инфекция, неонатальный гепатит, сепсис.
194

Наследственная тирозинемия I типа
Начальные проявления при 1Б типе обнаруживаются на первом году жизни в виде гепатомегалии, гипогликемии, потливости, слабости, полиурии, витамин- D-резистентного рахита.
Диагностика
Лабораторная диагностика
•Патогномоничным признаком НТ1 является высокий уровень сукцинилацетона в моче и плазме крови.
•Повышенное содержание ароматических аминокислот (тирозина, метионина, пролина, фенилаланина и др.).
•Альфафетопротеин (АФП) — маркер пролиферации желчных ходов, при НТ1Б повышен в десятки, а у детей с НТ1А даже в тысячи раз; повышение АФП — не специфичный, но чувствительный признак. При нормальном уровне АФП диагноз НТ1 сомнителен (у детей от 0 до 3 мес АФП < 1000 нг/мл; от 3 мес до 18 лет АФП < 12 нг/мл).
•Генетическое исследование является подтверждающим методом: обнаружение 2 мутаций (гомозиготы или компаунд-гетерозиготы) верифицирует диагноз.
Дифференциально-диагностический алгоритм
Симптом |
Возможный диагноз |
Гипертирозинемия |
Высокое содержание белка в рационе |
|
Тирозинемия II типа |
|
Тирозинемия III типа |
|
Другие болезни печени |
Гиперметионинемия |
Гомоцистинурия |
|
Нарушение метаболизма метионина |
|
Другие болезни печени |
|
Галактоземия тип 1, 3 |
|
Заболевание печени |
|
Непереносимость фруктозы |
|
Болезнь Нимана–Пика тип А/В |
|
Болезнь Вильсона |
|
Неонатальный гемохроматоз |
|
Гемофагоцитарный лимфогистиоцитоз |
|
Митохондриальная гепатопатия |
|
Врожденные нарушения гликозилирования |
|
Дефицит трансальдолазы |
|
Ацетаминофеновая интоксикация |
|
Бактериальные инфекции (сепсис, сальмонелез, туберкулез) |
|
Вирусные инфекции (CMV, гепатит А/В, герпес-вирусная |
|
инфекция) |
|
Отравление грибами (бледной поганкой) |
|
Лекарства из растительного сырья |
|
Идиосинкразия на различные агенты |
|
Инфильтративный или ишемический процесс |
Заболевание почек |
Синдром Лоу |
|
Цистиноз |
|
Почечный канальцевый ацидоз |
|
Фанкони синдром |
195
Атлас редких болезней
Симптом |
Возможный диагноз |
Рахит |
Гипофосфатазия |
|
Дефицит витамина D (нутритивный или генетически |
|
детерминированный) |
|
Гипофосфатемический рахит |
|
Фанкони синдром |
Неврологический |
Отек мозга, кровоизлияние |
кризис |
Вирусный/бактериальный менингит |
|
Гипернатриемическая дегидратация |
|
Острая перемежающаяся порфирия |
Лечение
Специфическая (субстрат-редуцирующая) терапия проводится нитизиноном (NTBC, 2-нитро-4-трифлюорометилбензоил-1,3-циклогександион), который не имеет аналогов, применяется в сочетании с низкобелковой диетой и назначением смесей, не содержащих тирозин и фенилаланин. Начальная доза 2 мг/кг, поддерживающая — в среднем 1 мг/кг.
Главное правило терапии нитизиноном (торговое название Орфадин, SOBI, Швеция) — непрерывность терапии, поскольку в ходе лечения концентрация тирозина повышается. Прекращение лечения приводит к образованию токсических метаболитов и тирозинемических кризов.
В Российской Федерации зарегистрированы 2 специализированных продукта на основе аминокислот без фенилаланина и тирозина: TYR Анамикс Инфант для детей от 0 до 12 мес в качестве основного источника питания и до 3-х лет в качестве дополнительного источника питания (состав приближен к грудному молоку, содержит LCP и пребиотики); XPHEN TYR Тирозидон для детей и взрослых (производитель «SHS International Ltd.» Великобритания, Nutricia Advanced Medical Nutrition).
Расчет лечебного питания (при терапии нитизиноном) для детей первого года жизни производят исходя из потребности в белке, близкой к физиологической (2,2–2,3 г/кг массы тела в сутки). Не менее 50–60% суточной потребности
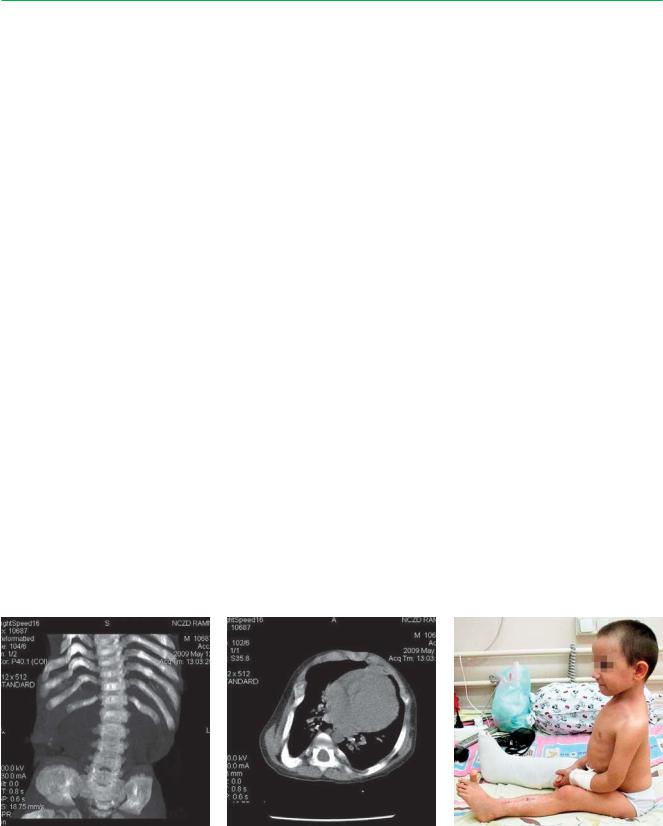
Рентгенограмма грудной клетки |
КТ грудной клетки пациента М., |
Никита М., 8 лет. Через 3 года |
позвоночника пациента М., 5 лет |
5,5 лет. Грубая деформация |
терапии нитизиноном и после |
(до лечения) |
грудной клетки, ребер, |
двух операций корригирующей |
|
позвоночника |
остеотомии, выполненной |
|
|
в травматолого-ортопедическом |
|
|
отделении Научного центра |
|
|
здоровья детей РАМН |
196

Наследственная тирозинемия I типа
в белке удовлетворяется за счет лечебной сухой смеси TYR Анамикс Инфант или Тирозидон, остальная часть (40–50%) компенсируется белком сцеженного материнского молока или детской молочной смесью с низким содержанием белка (1,2–1,3 г белка на 100 мл восстановленной смеси), а также низкобелковыми продуктами прикорма (овощными и фруктовыми пюре, безмолочными кашами с содержанием белка не более 0,5 г на 100 мл готового изделия, диетическими низкобелковыми продуктами Лопрофинами — макаронные изделия, заменитель яиц и муки, крекеры, рис, молочный напиток). Дефицит калорийности лечебного рациона компенсируется с помощью добавления жиров (растительного масла, но не более 3,5–4 г общего жира на 1 кг массы тела в сутки) и увеличения квоты углеводов за счет добавления вышеуказанных низкобелковых продуктов, а также 5% глюкозы. Аналогичным образом строится лечебный рацион у больных старше 1 года: общий белок рассчитывается исходя из безопасных потребностей в белке (не более 1,5–1,8 г/кг массы тела в сутки).
Прогноз. Без лечения умирает 90% детей в возрасте до 10 лет: чем раньше дебют заболевания, тем ниже уровень выживаемости. Причинами смерти от острой тирозинемии чаще являются катастрофическое кровотечение на фоне коагулопатии и острая печеночная недостаточность, при хронической форме — декомпенсированный цирроз печени и гепатоцеллюлярная карцинома. Риск развития последней у больных, не получающих субстрат-редуцирующей терапии, составляет 40%.
На фоне лечения прогноз хороший. Имеются публикации (2012) о положительном 20-летнем опыте применения нитизинона в мире: у больных НТ1 рождаются здоровые дети (гетерозиготные носители).
Список рекомендованной литературы
1.Баранов А.А., Намазова-Баранова Л.С., Сурков А.Н., Потапов А.С., Баканов М.И., Полякова С. И., Гундобина О. С., Лозоватор А. Л. Наследственная тирозинемия I типа: Учебное пособие. М.: ПедиатрЪ. 2012. 57 с.
2.URL: http://www.angis.org.au/bin/Databases/BIRX/birx_doc?phtomim+276700
3.Roth K.S. Tyrosinemia. 2006. URL: http://www.emedicine.com/ped/topic2339.htm
4.Mitchell G., Grompe M., Lambert M., Тanguay M. Hypertyrosinemia. In: Scriver C. The metabolic and molecular bases of inherited disease. New York, New York, USA: McGraw–Hill. 2001. Р. 1077–1106.
5.Scott C.R. The genetic tyrosinemias. Am J Med Genet. Part C. Seminars in medical Genetics. 2006; 142: 121–126.
6.Croffie J., Gupta S.K., Chong S.K.F., Fitzgerald J.F. Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics. 1999; 103 (3): 675–678.
197
Недержание пигмента
А |
Б |
В |
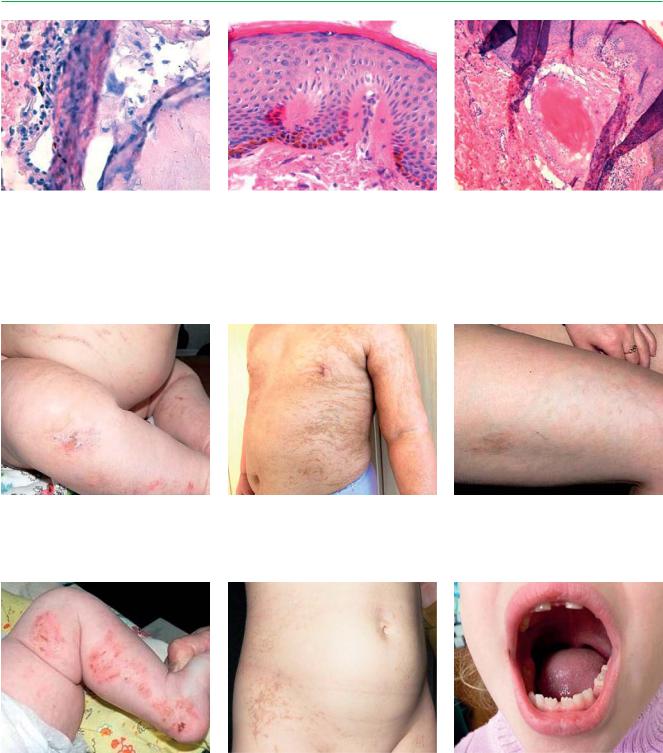
А–В. Гистология: В исследуемом препарате эпидермис с явлениями умеренного гиперкератоза, выраженного акантоза с неравномерно утолщенными и удлиненными акантолитическими выростами, очаговой вакуольной дистрофией. В эпидермоцитах базального слоя определяется большое количество пигмента. В дерме расположен крупный фокус некроза, окруженный инфильтратом из эпителиоидных клеток, лимфоцитов с примесью единичных гигантских многоядерных клеток типа «инородных тел». Морфологическая картина соответствует второй стадии синдрома Блоха–Сульцбергера
А |
Б |
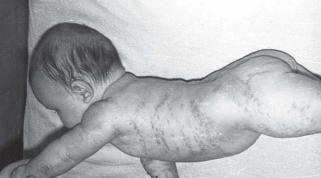
А, Б. Девочка с динамикой кожного процесса II и III стадии incontinentia pigmenti
Пациентка, 27 лет, атрофия и гипопигментация
(IV стадия incontinentia pigmenti)
А |
Б |
В |
А, Б. Девочка с динамикой кожного процесса I и III стадии. В. Неполный ряд зубов, дистрофия зубов, диастема
кожи — стадия I. Такие высыпания могут возникать на конечностях, реже они генерализованные. Иногда пузыри вскрываются, одновременно могут появляться новые высыпания. Также на конечностях и туловище могут возникать красные узелки или бляшки, возможно с изъязвлением. Лицо поражается редко. Такой характер высыпаний наблюдается в течение нескольких недель — нескольких месяцев, при этом может наблюдаться значительная эозинофилия.
199