

Аэробный гликолиз проходит 3 этапа:
Образование пирувата.
Окислительное декарбоксилирование пирувата (включает образование АцКоА).
Цикл трикарбоновых кислот (цикл Кребса), в который включается АцКоА и щавелево-уксусной кислоты (ЩУК), образуя лимонную кислоту. В конце из каждой молекулы глюкозы образуется 6 молекул СО2.
На этапах метаболизма глюкозы запускается цепь ферментов тканевого дыхания, первым из которых является НАД-зависимая дегидрогеназа. Дыхательная цепь срабатывает 10 раз за цикл, перенося 2Н+ на внешнюю сторону митохондриальной мембраны, а 2 электрона - на внутреннюю, что сопровождается выделением энергии, достаточной для синтеза 3-х молекул АТФ, т.е. всего 30 молекул АТФ за цикл.
Переход ФАД в ФАД Н2 дает укороченную цепь тканевого дыхания с 4-мя молекулами АТФ. Этот процесс, окислительное фосфорилирование сопряженное с дыханием, дает всего 34-е молекулы АТФ. Субстратное фосфорилирование еще 6 молекул АТФ.
Итого 40 молекул АТФ минус 2 молекулы (затраченные на фосфорилирование глюкозы) = 38 молекул АТФ и 6 молекул СО2 на 1-ую молекулу глюкозы. КПД около 44%.

Рис. 14.3. Цикл трикарбоновых кислот (цикл Кребса) [по Т.Т. Березову, Б.Ф. Коровкину, 1998].
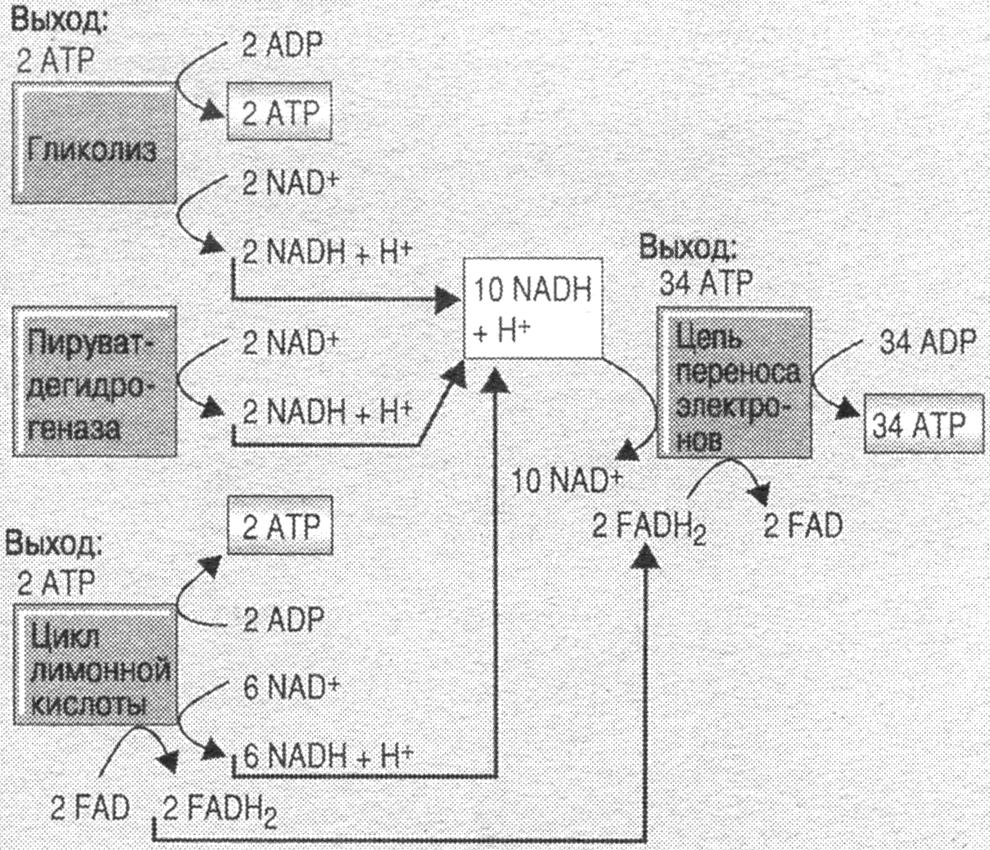
Рис. 14.4. Образование 38 молекул АТФ в результате окисления 1 молекулы глюкозы [по Б. Гринстейн, А. Гринстейн, 2000].
Анаэробный гликолиз совпадает до стадии пирувата. Далее вместо окислительного декарбоксилирования пируват подвергается восстановлению, приняв на себя 2Н+ от дегидрогенозы НАД Н2 с образованием лактата (молочной кислоты). Катализирует лактатдегидрогеназа. В эритроцитах только анаэробный гликолиз и большое значение в работающих мышцах. Эффективность - 2-е молекулы АТФ на 1-у молекулу глюкозы.
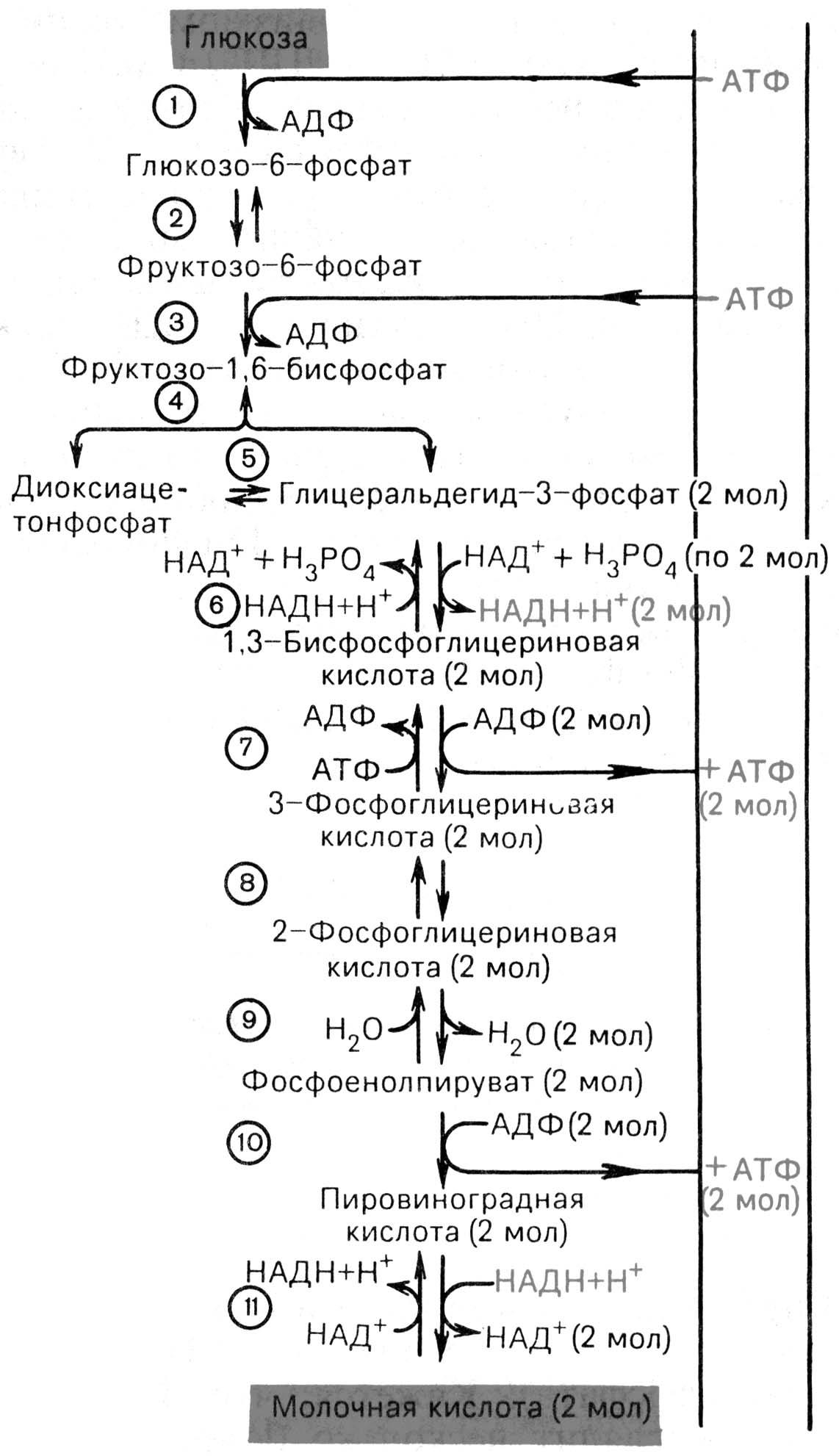
Рис. 14.5. Последовательность реакций гликолиза [по Т.Т. Березову, Б.Ф. Коровкину, 1998]. 1 - гексокиназа; 2 - фосфоглюкоизомераза; 3 - фосфофруктокиназа; 4 - альдолаза; 5 -триозофосфатизомераза; 6 - глицеральдегидфосфатдегидрогеназа; 7 -фосфоглицераткиназа; 8 - фосфоглицеромутаза; 9 - енолаза; 10 - пируваткиназа; 11 - лактатдегидрогеназа.
Пентозофосфатный путь - окислительная ветвь образует 2 молекулы НАДФ Н2 (необходимый для биосинтеза жирных кислот, холестерина и т.д.). В неокислительной ветви – рибозо-5-фосфат, который используется для синтеза РНК, ДНК, АТФ, КоА, НАД и ФАД.

Рис. 14.6. Схема пентозофосфатного пути метаболизма углеводов [по Т.Т. Березову, Б.Ф. Коровкину, 1998].
Глюконеогенез. Синтез глюкозы из неуглеводных продуктов; в первую очередь лактат и пируват, гликогенные аминокислоты, глицерол и ряд др. соединений. Т.е. предшественниками глюкозы может быть пируват или любое соединение, превращающееся в процессе катаболизма в пируват или один из промежуточных продуктов цикла Кребса.
Нарушения депонирования углеводов. После приема пищи большая часть глюкозы, метаболизирующейся в печени, превращается в гликоген, который при первой необходимости служит готовым источником глюкозы. Однако общее содержание его в печени довольно ограничено (в среднем 70-100 г) и способно обеспечить потребности организма в глюкозе в течение не более 8-12 часов.
Реакция образования гликогена зависит от активности гликогенсинтетазы, которая, в свою очередь, находится в обратной в зависимости от внутриклеточного уровня цАМФ.
До сих пор нет ясности в вопросе, опосредована ли активность гликогенсинтетазы главным образом гормонами (например, инсулином, глюкагоном или адреналином - первый ее повышает, два остальных - понижают) или субстратом, т.е. глюкозой.

Рис. 14.7. Синтез и распад гликогена [по Е.С. Северину, 2000]. 1, 2, 3, 4, 5 - реакции синтеза гликогена; 6, 7, 8 - реакции распада гликогена.
Снижение синтеза гликогена отмечается при миастении, гипоксии, тогда как повышенный распад наблюдается при охлаждении, перегревании, боли, судорогах, эмоциональном стрессе.
Выделяют т.н. агликогеноз - наследственное заболевание, вызванное дефектом гликогенсинтетазы. В печени почти или полностью отсутствует гликоген, выражена гипогликемия.
Следует сказать, что гликогенолиз контролируется ферментом фосфорилазой, которая, подобно гликогенсинтетазе, существует в неактивной форме и должна активироваться.
Гликогенолиз в печени и мышечной ткани приводит к образованию различных продуктов: в печени - к образованию свободной глюкозы, в мышцах - к высвобождению лактата и пирувата, поскольку глюкозо-6-фосфат не может превращаться в глюкозу, а вступает на гликолитический путь.
Гликогенолиз может рассматриваться как средство адаптации только к острым потребностям организма в глюкозе. В условиях длительного дефицита глюкозы (голодание, нарушение реабсорбции глюкозы в почках, диабет) в ход идет другой, более продолжительный механизм – глюконеогенез. Единственной другой тканью (помимо печени), в которой возможен глюконеогенез и превращение глюкозо-6-фосфата в глюкозу, является корковый слой почек.
Особую группу представляют болезни накопления гликогена, или гликогенозы. В основе этой патологии лежит энзиматический дефект, который проявляется необычной структурой гликогена или его избыточным накоплением в печени, почках, нервной системе. Характерны гепатоспленомегалия, задержка роста. Наиболее часто встречаются 6 типов гликогенозов.
I тип (гликогеноз Гирке) - следствие дефицита глюкозо-6-фосфатазы. Встречается наиболее часто, проявляется гипогликемией, накоплением гликогена в печени и почках, ацидозом (за счет накопления лактата) и гепатоспленомегалией. Больные отличаются малым ростом.
II тип (гликогеноз Помпе) - обусловлен дефектом кислой альфа-1,4-глюкозидазы. Отличается от других гликогенозов тем, что дефектным становится лизосомальный фермент. Проявляется генерализованным накоплением гликогена, поражением печени, почек, нервной системы, гипертрофией миокарда. Болезнь быстро прогрессирует и никакое лечение не в состоянии предотвратить смерть больного.
III тип (лимитдекстриноз, болезнь Кори, Болезнь Форбса) - вызывается дефицитом амило-1,6-гликозидазы. Больным свойственны гепатомегалия, мышечная, слабость, гипогликемия натощак, "кукольное личико". Течение относительно доброкачественное.
IV тип (амилопектиноз, болезнь Андерсена) - редко встречающаяся тяжелая форма гликогенозов. Для нее характерен цирроз печени с желтухой и печеночной недостаточностью, развивающийся в грудном возрасте. Отложение гликогена генерализованное, гликоген структурно изменен с очень длинными наружными ветвями. До сих пор не предложено никакого лечения, кроме симптоматического.
V тип (недостаточность миофосфорилазы, болезнь Мак-Ардла) - вызван дефицитом фосфорилазы, активирующей бета-киназу в мышцах и печени. Интересна история этого заболевания. Первый случай был расценен как психосоматическое нарушение. У больного в покое отсутствовали какие бы то ни было симптомы, но даже после умеренной нагрузки возникали боли в мышцах. Первые проявления болезни возникают обычно в 25-30 лет. Печень не поражается, структура гликогена нормальна, нет смертельных исходов, т.к. гамма-амилаза совместно с амило-1,6-гликозидазой расщепляют гликоген до глюкозы. Единственный признак - миастения, особенно после физической активности.
VI тип (недостаточность печеночного фосфорилазного комплекса, болезнь Херса) - дефект печеночной фосфорилазы, ведущий и избыточному накоплению нормального гликогена в печени. Отмечают гепатомегалию, легкое замедление темпов роста. Прогноз для жизни хороший, умственное развитие нормальное.
Достаточно редко встречается гликогеноз VII типа (дефект мышечной фосфофруктокиназы, болезнь Терье), схожий с болезнью Мак-Ардла и проявляющийся нарастанием уровня лактата и пирувата в крови после мышечной работы.
Этиология и патогенез гипогликемических состояний.