

Gallup G.A. - Valence Bond Methods, Theory and applications (CUP, 2002)
.pdf224 16 Interaction of molecular fragments
|
|
|
Table 16.7. |
The leading HLSP functions for the ground state wave function |
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
of C |
3 H6 |
at the equilibrium geometry when hybrid orbitals are used. |
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
2 |
|
|
|
|
|
3 |
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Num. |
|
|
|
|
1 |
|
|
|
|
|
6 |
|
|
|
|
6 |
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
h |
32 |
h |
11 |
|
|
h |
11 |
h |
11 |
|
h |
11 |
h |
11 |
|
h |
11 |
h |
11 |
|
|
|
|
|
Tab. |
|
|
|
h |
12 |
h |
21 |
h |
12 |
h |
21 |
h |
31 |
h |
31 |
|
h |
21 |
h |
21 |
|||||
|
|
|
|
|
|
|
h |
22 |
h |
31 |
R |
|
h |
22 |
h |
31 |
R |
h |
12 |
h |
21 |
R |
h |
31 |
h |
31 |
R |
|
|
|
C i (min |
) |
|
|
|
0.354 55 |
|
0.098 99 |
|
0.052 40 |
|
0.046 07 |
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
of |
σi and |
πi |
orbitals is not |
very easily interpreted, although the |
leading |
term, in |
|
|
|
|
|
|
|
|||||||||||||||
the case of HLSP functions, is the same as that for the ground state. We do not |
|
|
|
|
|
|
|
|||||||||||||||||||||
give them here, but the first excited state in terms of the hybrid orbitals is likewise |
|
|
|
|
|
|
|
|
||||||||||||||||||||
poorly illuminating. We may look at the problem in another way. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
As cyclopropane dissociates, we see that the geometry changes happen rather |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
rapidly over a fairly narrow range as the character of the energy states changes in |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
the |
neighborhood |
of |
|
|
R |
1 = 2.4 A |
|
. (See |
|
Fig. 16.4.) |
At |
asymptotic |
geometries |
we |
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||||||
saw that the characters of the wave |
functions for the first two states are |
clearcut. |
|
|
|
|
|
|
|
|
||||||||||||||||||
As the one methylene moves, the two pieces in the first excited state, consisting of |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
two triplet fragments, attract one another more strongly and the potential energy |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
curve falls, see Fig. 16.3. The ground state, consisting of two singlet fragments |
|
|
|
|
|
|
|
|
||||||||||||||||||||
appears repulsive. These two sorts of states would cross if they did not interact. |
|
|
|
|
|
|
|
|
||||||||||||||||||||
They, in fact, do interact: there is an avoided crossing, and a barrier appears on the |
|
|
|
|
|
|
|
|
||||||||||||||||||||
lower curve. This interaction region is fairly narrow, and, inside the cross-over, the |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
lower curve continues downward representing the |
bonding |
that |
holds |
C |
|
|
|
|
|
|
|
|
3 H 6 |
|||||||||||||||
together. Thus, this targeted correlation treatment predicts that there is a 1.244 eV |
|
|
|
|
|
|
|
|
||||||||||||||||||||
barrier to the insertion of singlet |
methylene into ethylene to form cyclopropane. |
|
|
|
|
|
|
|
|
|
||||||||||||||||||
We do not show it here[39], but triplet methylene and singlet ethylene repel each |
|
|
|
|
|
|
|
|
||||||||||||||||||||
other strongly at all distances, and thus should not react unless there should be a |
|
|
|
|
|
|
|
|
||||||||||||||||||||
spin cross-over to a singlet state. This occurrence of a barrier due to an avoided |
|
|
|
|
|
|
|
|
||||||||||||||||||||
crossing has been invoked many times to explain and rationalize reaction pathways |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
[67, 69]. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
16.1.4 |
Cyclopropane with an STO-3G basis |
|
|
|
|
|
|
|
|
|
|
|
|||||||||
Some years ago a short description of a more restricted version of the problem in |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
the last section was published[39]. Using an STO-3G basis, the earlier calculation |
|
|
|
|
|
|
|
|
||||||||||||||||||||
examined the two lowest |
|
|
1 A 1 energies as a singlet methylene approached an ethy- |
|
|
|
|
|
|
|||||||||||||||||||
lene molecule. In this case, however, the ethylene was not allowed to relax in its |
|
|
|
|
|
|
|
|
||||||||||||||||||||
geometry. The |
curves |
are |
shown |
in Fig. 16.5. The |
important |
point |
is that |
we |
see |
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
16.2 |
Formaldehyde, H |
|
2CO |
|
225 |
|
14 |
|
|
|
|
|
|
|
|
12 |
|
|
|
|
|
|
|
|
10 |
21A |
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(eV) |
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Energy |
6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 |
11A |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
|
1 |
2 |
|
3 |
|
4 |
5 |
6 |
|
|
|
|
R |
1 |
(Å) |
|
|
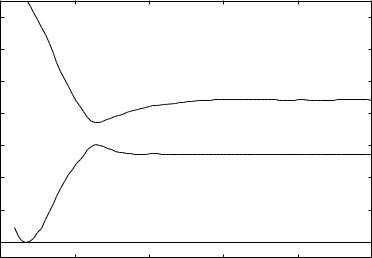
Figure 16.5. The two lowest 1 A 1 states showing the attack of singlet methylene on a rigid ethylene. These energies were obtained using an STO-3G basis, with which we obtain a
barrier of about 0.8 eV.
the same qualitative behavior in this much more approximate calculation as that shown in Fig. 16.3, where the results using a larger basis and fuller optimization is presented.
|
|
16.2 Formaldehyde, H |
|
2 CO |
|
When formaldehyde is subjected to suitable optical excitation it dissociates into H |
2 |
||||
and CO. The process is thought to involve an excitation to the first excited singlet |
|
||||
state followed by internal conversion to a highly excited vibrational state of the |
|
||||
ground singlet state that dissociates according to the equation |
|
|
|
||
|
H |
h ν |
→ H 2 |
+ CO. |
|
|
2CO → H 2CO |
|
|||
Conventional counting says that H |
|
2CO has four bonds in it, and the final product has |
|
||
the same number arranged differently. Our goal is to follow the bonding arrange- |
|
||||
ment from the initial geometry to the final. This is said to occur on the |
|
S 0 (ground |
|||
state singlet) energy surface, which in full generality depends upon six geometric |
|
||||
parameters. Restricting the surface to planar geometries reduces this number to |
|
||||
five, and keeping the C—O distance fixed reduces it to four. We will examine |
|
||||
different portions of the |
S 0 |
surface for different numbers of geometric coordinates. |
|
||
Some years ago Vance and the present author[68] made a study of this surface |
|
||||
with the targeted correlation technique using a Dunning double- |
|
zeta |
basis[70] that, |
||
226 16 Interaction of molecular fragments
|
|
Table 16.8. |
Orbitals used and statistics of MCVB calculations on the |
S |
|
|
0 |
||||||
|
|
energy surface of formaldehyde. For the 6-31G |
|
|
|
basis the full set of |
|
||||||
|
|
configurations from the unprimed orbitals was used and |
|
|
|
|
|
||||||
|
|
|
single excitations into the primed set were included. |
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
Orbitals |
|
|
Total |
|
C 2v |
C s |
||
|
STO3G basis |
H:1 |
s a |
, 1s b |
|
|
|
|
|
|
|
|
|
|
|
|
|
CO:3 |
σ, 3 σ, 5σ, 6 σ, 1π, 2π |
|
1120 |
565 |
1120 |
||||
|
6-31G* basis |
H:1 |
s a |
, 1s b , 2s a , 2s b |
|
|
131 |
|
70 |
131 |
|||
|
|
|
|
CO:5 σ, 1π, 5σ , 2π, 2π |
|
|
|||||||
|
|
|
|
|
|
||||||||
except for the lack of polarization functions, is similar to a 6-31G |
|
|
|
basis. To keep |
|||||||||
consistency with the remainder of this book we redo some of the calculations from |
|
|
|
|
|
||||||||
the earlier study with the latter basis, but will mix in some of the earlier results, |
|
|
|
||||||||||
which are essentially the same, with the current ones. |
|
|
|
|
|
|
|
|
|||||
We show the results of calculations at the STO3G and 6-31G |
|
|
|
levels of the AO |
|||||||||
basis. Table 16.8 shows the orbitals used and the number of functions produced for |
|
|
|
|
|||||||||
each case. These statistics apply to each of the calculations we give. |
|
|
|
|
|
|
|||||||
The important difference between the STO3G and 6-31G |
|
|
|
bases is the arrange- |
|||||||||
ment of orbitals on the CO fragment. In its ground state CO has an orbital config- |
|
|
|
|
|||||||||
uration of |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
Core: 3 |
σ 24σ 25σ 21π 4. |
|
|
|
|
|||
The 5 |
σ function |
is best described as a nonbonding orbital located principally on |
|
|
|
||||||||
the C atom. In Table 16.8 the 2 |
|
π orbital is the virtual orbital from the ground state |
|
||||||||||
RHF treatment. The |
primed orbitals on H are the same as we have used before, |
|
|
|
|
||||||||
but those on CO are |
based upon an |
ROHF |
|
|
n |
→ π calculation of the |
first triplet |
||||||
state. The “raw” 5 |
σ , 5σ , 2π , and 2 |
π taken |
directly |
from |
the calculations |
will |
|
||||||
not work, however. Their overlaps are much too large for an |
|
|
|
|
S matrix of any size |
||||||||
(>2 or 3) to be considered nonsingular by standard 16-place accuracy calculations. |
|
|
|
||||||||||
Therefore, for each high-overlap pair the sum and difference were formed. These |
|
|
|
|
|
||||||||
are orthogonal, and do not cause any problems. |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
16.2.1 The least motion path |
|
|
|
|
|
|
|||
We first |
comment on |
the so-called |
least motion path |
(LMP), in which |
the |
two |
|
|
|
||||
H atoms move away from the CO atoms, maintaining a |
|
|
|
|
C |
2v symmetry, as |
shown |
||||||
in Fig. 16.6. Earlier calculations of all |
sorts indicate |
that this |
path does |
not |
cross |
|
|
|
|||||


16.2 Formaldehyde, H 2CO 229
Table 16.11. |
|
The leading terms in the wave function for the equilibrium geometry |
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
of formaldehyde. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
2 |
|
|
|
|
3 |
|
|
|
|
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Standard |
|
Num. |
|
|
|
1 |
|
|
|
|
|
|
2 |
|
|
|
|
2 |
|
|
|
|
|
2 |
|
|
tableaux |
|
|
|
1πx |
1πx |
|
5σ |
5σ |
|
|
1πx |
1πx |
|
|
1πx |
1πx |
|
|||||||||
functions |
|
Tab. |
1πy |
1πy |
|
1πx |
1πx |
1πy |
1πy |
1πy |
1πy |
|
||||||||||||||
|
|
|
|
1s b |
2π |
|
|
1s a |
2π |
|
1s b |
5σ |
|
2π |
1sa |
|
||||||||||
|
|
|
|
1s a |
5σ |
|
|
|
1πy |
1πy |
|
2πy |
2πy |
|
1s b |
5σ |
|
|||||||||
|
|
|
|
|
|
y |
|
y |
|
|
|
y |
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
C |
i (min) |
|
|
0.296 23 |
|
|
0.178 83 |
|
0.136 65 |
|
|
0.119 13 |
|
|
|
||||||||||
HLSP |
|
Num. |
|
|
|
1 |
1πy |
|
|
|
1πx |
2 |
|
|
1πy |
2 |
|
|
|
1πy |
2 |
|
||||
functions |
|
Tab. |
1πy |
|
1πx |
1πy |
1πy |
|||||||||||||||||||
|
|
|
1πx |
1πx |
|
|
5σ |
5σ |
|
|
1πx |
1πx |
|
|
1πx |
1πx |
|
|||||||||
|
|
|
1s a |
|
2π |
|
1s b |
2π |
2π |
1sb |
|
1s b y |
5σ |
|||||||||||||
|
|
|
|
1s b |
|
5σ |
|
R |
|
1πy |
1πy |
R |
|
1s a |
5σ |
|
R |
|
2π |
2πy |
R |
|||||
|
|
|
|
|
|
y |
|
|
|
y |
|
y |
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
i (min) |
|
|
0.310 08 |
|
|
0.178 83 |
|
0.159 18 |
|
|
|
|
−0.136 65 |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
1. The leading |
term represents a structure with |
|
|
|
|
|
|
an electron pair |
bond between one |
H and |
|
|
|
|
|
|
||||||||||
the 5σ |
orbital and another between the other H and the 2π |
|
|
|
|
y |
orbital. |
|
|
|
|
|
|
|
||||||||||||
2. This term together with the first provides |
the two Rumer diagrams for the bonding |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
scheme. That this has such a large coefficient indicates that electron pair bonds are not |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
near perfect pairing. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
3. This term involves correlation and polarization on the CO portion of the |
|
|
|
|
|
|
|
|
system with the |
|
|
|||||||||||||||
electron pair bonds to the Hs still in place. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
4. The fourth term involves a further rearrangement of the electrons on the CO portion of |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
the system. In this case one H is now bonded to the 1 |
|
|
|
|
|
πy instead of the 2 |
πy |
orbital. These |
|
|
||||||||||||||||
terms provide correlation, polarization, and also give a combination of both bonding and |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
antibonding |
|
πy orbitals |
so that this sort of |
bond |
|
will disappear |
and |
C—H |
bonds and |
|
|
|
|
|
|
|
|
|||||||||
O nonbonding orbitals will appear as the molecule forms. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
Table 16.11 shows the leading terms in the wave function at the equilibrium |
|
|
|
|
|
|
|
|
|
|||||||||||||||||
geometry of H |
|
2CO in both standard tableaux function and HLSP function form. |
|
|
|
|
|
|
|
|
||||||||||||||||
1. The first standard |
tableaux |
function |
term |
is essentially triplet H |
|
|
|
|
|
2 (much |
elongated, |
of |
|
|
||||||||||||
course) coupled with the |
|
|
3 |
y |
state of CO. The HLSP function has the same interpretation. |
|
|
|
|
|
|
|||||||||||||||
2.The second terms are the same and are an ionic type associated with the first term. These provide delocalization.
3.The third standard tableaux function and fourth HLSP function terms are the same. These
are both ionic and provide antibonding character in the |
y -direction to “remove” that part |
of the original triple bond in CO. |
|
4.The fourth standard tableaux function term and the third HLSP function term are the same configuration but not the same function. In both cases, however, the terms involve
breathing for the 1s |
orbitals in the H atoms. |
232 |
|
|
|
|
|
References |
|
|
|
|
|
|
||
[27] |
J. C. Slater. |
Quantum Theory of Molecules and Solids |
|
|
|
|
|
, Vol. 1. McGraw-Hill Book |
|
|||||
|
|
Co., New York (1963). |
|
|
|
|
|
|
|
|
|
|
||
[28] |
M. Abramowitz and I. A. Stegun. |
|
Handbook of Mathematical Functions |
. National |
||||||||||
|
|
Bureau of Standards, US Government Printing Office (1970). |
|
|
|
|
||||||||
[29] |
S. C. Wang. |
Phys. Rev. |
, 31 |
, 579 (1928). |
|
|
|
|
|
|
||||
[30] S. Weinbaum. |
|
J. Chem. Phys |
, 1 , 593 (1933). |
|
|
|
||||||||
[31] |
W. Kolos and L. Wolniewicz. |
|
J. Chem. Phys. |
|
, 41 , 3663 (1964). |
|
||||||||
[32] |
N. Rosen. |
Phys. Rev. |
, 38 , 2099 (1931). |
|
|
|
|
|
|
|||||
[33] S. F. Boys. |
Proc. Roy. Soc. (London) |
, A200 |
|
, 542 (1950). |
|
|||||||||
[34] |
G. G. Balint-Kurti and M. Karplus. |
|
J. Chem. Phys. |
|
, 50 |
, 478 (1968). |
|
|||||||
[35] G. Wannier. |
Phys. Rev. |
, 52 |
, 191 (1937). |
|
|
|
|
|
|
|||||
[36] |
R. Pauncz. |
The Construction of Spin Eigenfunctions |
|
|
|
|
. Kluwer Academic/Plenum, |
|
||||||
|
|
New York (2000). |
|
|
|
|
|
|
|
|
|
|
||
[37] |
D. E. Littlewood. |
The Theory of Group Characters. |
|
|
|
|
Oxford University Press, London |
|||||||
|
|
(1950). |
|
|
|
|
|
|
|
|
|
|
|
|
[38] |
H. Weyl. |
The Theory of Groups and Quantum Mechanics |
|
|
|
|
|
. Dover Publications, Inc., |
||||||
|
|
London (1931). |
|
|
|
|
|
|
|
|
|
|
, 16 , |
|
[39] |
G. A. Gallup, R. L. Vance, J. R. Collins, and J. M. Norbeck. |
|
|
|
|
Ad. Quantum Chem. |
||||||||
|
|
229 (1982). |
|
|
|
, 1 , 60 (1964). |
|
|
|
|||||
[40] |
F. A. Matsen. |
Ad. Quantum Chem. |
, 157 |
|
|
|
||||||||
[41] |
W. A. Goddard III. |
|
Phys. Rev. |
, 81 (1967). |
|
|
|
|
||||||
[42] |
A. C. Aitken. |
|
Determinants and Matrices |
|
|
. Interscience Publishers, New York (1956). |
||||||||
[43] |
G. A. Gallup. |
|
Intern. J. Quantum Chem. |
|
, 16 |
, 267 (1974). |
|
|||||||
[44] |
|
P. C. Hiberty. |
|
THEOCHEM |
, 398–399 |
, 32 |
, 35 (1997). |
|
|
|||||
[45] |
C. C. J. Roothaan. |
|
Rev. Mod. Phys. |
, 179 (1960). |
|
|
||||||||
[46] |
|
M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. |
|
|
||||||||||
|
|
Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis, |
|
|||||||||||
|
|
and J. A. Montgomery. |
J. Comput. Chem. |
|
, 14 |
, 1347 (1993). |
|
|||||||
[47] |
|
D. Chen and G. A. Gallup. |
|
J. Chem. Phys. |
|
, 93 |
, 8893 (1990). |
|
||||||
[48] |
S. Huzinaga. |
|
Gaussian Basis Sets for Molecular Calculations |
|
|
|
. Elsevier Science |
|||||||
|
|
Publishing Co., New York (1984). |
|
|
|
|
|
|
|
|
|
|||
[49] |
|
K. P. Huber and G. Herzberg. |
|
Molecular Spectra and Molecular Structure. IV. |
|
|||||||||
|
|
Constants of Diatomic Molecules. |
|
Van Nostrand Reinhold, New York (1979). |
|
|||||||||
[50] |
L. Pauling. |
The Nature of the Chemical Bond |
|
|
|
|
. Cornell University Press, Ithica |
|
||||||
|
|
(1960). |
|
|
|
, 2 , 782 (1934). |
|
|
|
|||||
[51] |
R. S. Mulliken. |
|
J. Chem. Phys. |
|
|
|
||||||||
[52] L. Allen. |
J. Amer. Chem. Soc. |
, 111 |
, 9003 (1989). |
|
, 14 , 1440 (1993). |
|||||||||
[53] S. Huzinaga, E. Miyoshi, and M. Sekiya. |
|
|
|
J. Comp. Chem. |
|
|||||||||
[54] |
G. Herzberg. |
|
Molecule Spectra and Molecular Structure. III. Electronic Spectra and |
|
||||||||||
|
|
Electronic Structure of Polyatomic Molecules. |
|
|
|
|
Van Nostrand Reinhold, New York |
|
||||||
|
|
(1966). |
|
|
|
, 4 , 581 (1936). |
|
|
|
|
|
|||
[55] H. H. Voge. |
J. Chem. Phys. |
|
|
|
|
|
||||||||
[56] |
A. Veillard. |
Theoret. Chim. Acta |
, 18 , 21 (1970). |
|
|
|
||||||||
[57] |
V. Pophristic and L. Goodman. |
Nature |
, 411 |
, 565 (2001). |
|
|||||||||
[58] |
R. B. Woodward and R. Hoffman. |
|
|
The Conservation of Orbital Symmetry |
. Academic |
|||||||||
|
|
Press, New York (1970). |
|
|
|
|
|
|
|
|
|
|
||
[59] |
M. Randi´ |
c. in Valence Bond Theory and Chemical Structure. |
|
|
|
|
Ed. by D. J. Klein and |
|||||||
|
|
N. Trinajsti´c. Elsevier, Amsterdam (1990). |
|
|
|
|
, 96 , 3386 (1974). |
|
||||||
[60] |
J. M. Norbeck and G. A. Gallup. |
|
J. Amer. Chem. Soc. |
|
|
|||||||||
[61] |
D. L. Cooper, J. Gerratt, and M. Raimondi. |
|
|
|
Nature |
, 323 |
, 699 (1986). |
|
||||||
|
|
|
|
References |
|
233 |
[62] |
S. Shaik, A. Shurki, D. Danovich, and P. C. Hiberty. |
J. Mol. Struct. (THEOCHEM), |
||||
|
|
398, 155 (1997). |
|
|
|
101, 1501 (2001). |
[63] |
S. Shaik, A. Shurki, D. Danovich, and P. C. Hiberty. |
Chem. Rev. |
||||
[64] |
|
R. S. Mulliken and R. G. Parr. |
J. Chem. Phys., |
19, 1271 (1951). |
|
|
[65] |
H.-D. Beckhaus, R. Faust, A. J. Matzger, D. L. Mohler, D. W. Rogers, C. R¨uchart, |
|
||||
|
|
A. K. Sawhney, S. P. Verevkin, K. P. C. Vollhardt, and S. Wolff. |
|
J. Amer. Chem. Soc., |
||
|
|
122, 7819 (2000). |
|
|
104, 1530 (1982). |
|
[66] |
|
J. R. Collins and G. A. Gallup. |
J. Amer. Chem. Soc., |
|||
[67] |
|
S. Shaik and A. Shurki. |
Angew. Chem. Int. Ed., |
38, 586 (1999). |
|
|
[68] |
R. L. Vance and G. A. Gallup, Chem. Phys. Lett., |
81, 98 (1981). |
|
|||
[69] |
|
S. S. Shaik, H. B. Schlegel, and S. Wolfe. |
Theoretical Aspects of Physical Organic |
|||
|
|
Chemistry. The S |
N2 Mechanism. Wiley and Sons Inc., New York (1992). |
|
||
[70] |
|
T. H. Dunning. |
J. Chem. Phys., |
65, 2823 (1970). |
|
|
[71] |
W. J. Hehre, L. Radom, P. v.R. Schleyer, and J. A. Pople. |
Ab Initio Molecular Orbital |
||||
|
|
Theory. Wiley and Sons, New York (1986). |
|
|
|
|