

19. Rearrangement reactions involving the amino, nitro and nitroso groups 867
+ O− |
X |
+ |
O− |
|
|
N |
N |
|
|
|
|
N |
X |
|
N |
|
|
(30) |
|
(31) |
|
|
|
+ |
|
|
− |
|
|
OH |
|
+ |
O |
|
|
N |
|
N |
|
|
|
N |
X |
|
N |
|
N |
(32) |
|
(33) |
|
|
|
|
F |
F |
|
|
|
N + |
|
+ |
O− |
F |
F |
|
N |
N |
|
|
|
N |
N |
F |
|
|
|
−O |
F |
|
|
|
|
|
|
|
F |
F |
|
|
|
|
|
||
(34) |
|
(35) |
|
|
|
+ +
Ar N N Ar). A number of possible reaction mechanisms have been suggested at different times and, at this time, the position is not settled. No heavy-atom KIE work has been reported for the acid catalysed reaction, but such experiments have been carried out for the photochemical reaction37 which gives the 2-hydroxy product and which is known to be intramolecular. There is an absence of a KIE when [15N,15N0 ] material is used, which at least reveals that if the proposed intermediate 36 is involved, then the rate-limiting step must be its formation and not its subsequent reaction since N O bond fission cannot be part of the slow step.
N
N
O
H
(36)
IV. REARRANGEMENT INVOLVING PHENYLHYDROXYLAMINES
A. The Bamberger Rearrangement
This is the best known rearrangement reaction of phenylhydroxylamines and is an acid catalysed reaction leading principally to the formation of 4-amino phenols 37, although a little of the 2-isomers 38 are also sometimes formed. Reaction proceeds quite smoothly in relatively dilute acid at room temperature. Reaction is quite general for a range of R and X substituents. Much of the early work was carried out by Bamberger38 and the position up to 1967 has been very well reviewed39.

868 |
D. Lyn H. Williams |
|
|
PhNHOH |
RNH |
RNH |
OH |
|
H+ |
+ |
|
|
|
|
|
X |
X OH |
|
X |
|
(37) |
|
(38) |
In the presence of alcohols, the corresponding ethers are formed and added nucleophiles such as chloride ion40 or azide ion41 lead to the chloroand azido-amine products, respectively. Rate constants are independent of the concentration of added nucleophile. Labelled 18O from the solvent is incorporated in the product42. All the evidence points to a reaction mechanism where water is lost from the O-protonated reactant to give a nitrenium ion iminium ion intermediate which is rapidly trapped by a nucleophile (H2O in this case) to give the final product. This is shown in Scheme 7. Protonation at N- is likely to be more extensive, but there is no pathway to products from the N-protonated intermediate.
H2 N+OH
HNOH
H+
H |
+ |
+ |
HN+ |
|
|
|
|
|
NH |
||
|
|
HNOH2 |
NH |
||
|
|
|
Slow |
|
+ |
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
Fast H2 O |
|
|
|
|
NH2 |
NH2 |
|
|
|
|
|
|
OH |
|
|
|
|
+ |
|
OH
SCHEME 7

19. Rearrangement reactions involving the amino, nitro and nitroso groups 869
6 + log kobs
2.5
2.0
H2SO4− H2O
1.5
D2SO4− D2O
1.0 


|
|
|
|
|
|
|
|
− |
|
1 |
− |
|
2 |
− |
|
3 |
− |
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||
2 |
1 |
0 |
|
|
|
|
|||||||||||||
|
|
|
pH |
|
|
|
|
H0 |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
FIGURE 1. Plot of log kobs vs H0 or pH H2SO4 H2O and in D2SO4D2O. Reproduced
Reference 44
for the rearrangement of N-phenylhydroxylamine in by permission of the Royal Society of Chemistry from
More recent mechanistic studies43,44 have confirmed the general mechanistic framework. Acid catalysis is found at acidities up to ca pH 1, then there is an acid region where the rate constant is acid-independent, then at higher acidities acid catalysis occurs again. This is shown in Figure 1.
The plateau region corresponds to effectively complete N-protonation. The pKa value measured spectrophotometrically (1.90) agreed with that derived from the kinetic measurements. Similar good agreement was obtained for the N-Et and 4-Me reactants and also for the unsubstituted phenylhydroxylamine in D2O. The measured solvent KIE was also
in agreement with the mechanism in Scheme 7. Acid catalysis at high acidity is believed
+
to arise from another reaction pathway involving the doubly protonated species Ph NH2
+
OH2 for which there is support from polarographic measurements at high acidity45. Kinetic experiments using 3-ring substituted derivatives43 gave a good log k vs m
correlation yielding a value of 3.2, confirming N O bond fission in the sense leading to positive charge increase on the nitrogen atom in the transition state.
N-Ethyl substitution had very little effect on the measured rate constant, whereas a 4-methyl substituent increased the rate constant by a factor of ca 100. In this case the initial product (identified by Bamberger) is the iminocyclohexadienol 39, which slowly hydrolyses to the quinone 40. These substituent effects suggest that in the transition state the developing positive change is located mostly at the 4-position (stabilized by the 4-Me substituent) and very little on the nitrogen atom (no stabilization by a N-Et substituent), so that the intermediate is more properly described by the iminium ion. This is supported by an earlier observation46 that whilst full incorporation of 18O from the solvent H218O occurs in the product, there is no detectable 18O incorporation into the reactant phenylhydroxylamine.

870 |
D. Lyn H. Williams |
||
HNOH |
NH |
|
O |
|
H+ |
Slow |
|
|
|
H2O |
|
Me |
Me OH |
|
Me OH |
|
|
|
|
|
(39) |
(40) |
|
Further kinetic experiments with some sterically hindered phenylhydroxylamines gave results47 which suggest that under certain circumstances steric acceleration occurs, attributable to the buttressing effect of neighbouring 3-substituents. Thus the rate constants for the reactions of 41 and 42 are respectively greater than are those for 43 and 44.
HNOH |
HNOH |
HNOH |
|
HNOH |
Me(or Hal) |
|
Me |
Me |
Me |
|
|
Me |
Me |
|
(41) |
(42) |
(43) |
|
(44) |
The rearrangement reaction continues to be of synthetic utility, often involved in industrial processes. Patent references (e.g. Reference 48) refer to the formation of 4-amino phenols. Often the reactant nitro compound is reduced (to the hydroxylamine) in an acid environment so that the two-stage reaction can be accomplished as a one-pot synthesis. 4-Amino phenol itself 45 can be made in high yield directly from nitrobenzene49 and the 4-methoxy aniline derivative 46 similarly from 2-methylnitrobenzene by hydrogenation in MeOH/H2SO450.
NO2 |
NH2 |
Catalytic hydrogenation
H+
OH
(45)
Me |
NO2 |
Me |
NH2 |
Hydrogenation
MeOH/H2SO4
MeO
(46)

19. Rearrangement reactions involving the amino, nitro and nitroso groups 871
At low acidities oxidation of phenylhydroxylamine occurs yielding azoxybenzene and other products. This competing reaction can be eliminated by working anaerobically and can also be much reduced by working at high acidities, suggesting that the oxidation occurs via the free base form of phenylhydroxylamine.
An unusual kinetic result has been reported51 when phenylhydroxylamine reacted anaerobically with bisulphite anion. The product distribution was as expected, i.e. both 2- and 4-aminophenol and the 2- and 4-aminobenzenesulphonates were formed. Kinetic measurements showed a first-order dependence upon [bisulphite], in contrast to the earlier work with Cl and later with N3 . The authors propose a mechanism involving direct attack by the nucleophile at the 2- and 4-positions as the rate-limiting step, followed by proton transfers and solvent attack to form the sulphonate products.
There has been considerable interest in the chemistry of hydroxylamines, since it is believed52 that the carcinogenicity of some arylamines results from the formation of the N-hydroxy species, which in turn generate nitrenium ions that react in a conventional electrophilic sense with nucleic acids.
B. Other Rearrangements
A different mechanism probably operates for the reaction of N-hydroxy-N- phenylamides in the presence of (n-Bu)3P, CCl4 and MeCN, with the 2-isomer in the product53 suggestive of some intramolecular pathway as outlined in Scheme 8.
O |
O |
|
|
O |
||
|
|
|
||||
|
|
RC |
|
O |
|
|
RCNOH |
N |
P(n−Bu)3 |
RCNH |
|||
|
|
|
|
|
|
+ |
|
|
|
|
|
|
OP(n−Bu)3 |
|
(n-Bu)3 P |
|
|
|
|
|
|
CCl4 , MeCN |
|
|
|
|
|
O
RCNH
OH
SCHEME 8
O-Substituted phenylhydroxylamines also undergo rearrangement to give the 2-isomers. For example O-(arenesulphonyl) phenylhydroxylamines 47 readily form the 2-sulphonyl derivatives 48. Experiments with 18O-labelled compounds led to the suggestion54 of a mechanism involving an ion pair which has only a very short lifetime.
Heating O-phenylhydroxylamine 49 gave 2-aminophenol, though not in very high yield55. A detailed mechanistic investigation of this reaction in trifluoroacetic acid has

872 |
D. Lyn H. Williams |
PhCONOSO2 R PhCONH
OSO2 R
(47) |
|
(48) |
|
ONH2 |
OH |
OH |
|
|
|
|
NH2 |
|
TFA |
|
+ |
|
|
|
|
NH2
(49) |
50% |
7% |
been carried out56. A little of the 4-isomer is formed, but the predominance of the 2- isomer suggests that this reaction is very different from the Bamberger rearrangement. Cross-coupling experiments with 15N labelled compounds showed that the 2-isomer is formed in an intramolecular process and that the formation of the 4-isomer has both intramolecular and intermolecular reaction pathways. Substituent effects in the aromatic ring gave a large negative value ( 7.8) from a Hammett plot using C values, indicating that a positive charge is being generated on the oxygen atom (delocalized into the ring to a considerable degree) in the transition state. The suggested mechanism outlined in Scheme 9 involves N-protonation followed by the formation of a tight phenoxenium
|
+ |
|
ONH2 |
|
ONH3 |
|
H + |
|
|
|
|
|
|
|
|
Slow |
||
OH |
|
|
O |
|
|
OH |
|
|
|
|
|||
|
NH2 |
|
|
|
||
|
intramolecular |
|
|
intramolecular |
||
+: NH3
NH2
(50)
SCHEME 9

19. Rearrangement reactions involving the amino, nitro and nitroso groups 873
ion ammonia pair (50) which can collapse to give the products. Reaction of the solvent (TFA) with the ion pair gives a solvent separated ion pair from which it is possible to rationalize the formation of the two minor by-products, catechol and hydroquinone.
A synthetic application of this reaction has been reported57 when the rearrangement of 2-aryl-O-phenylhydroxylamines is followed by a ring enlargement to give an aryldihydroazepinone (Scheme 10). The 2-aryl-2-phenyl intermediate was also trapped out as the N-trifluoroacetamide.
ONH2 |
+ |
O |
|
ONH3 |
|||
|
|||
|
+ |
: NH3 |
|
Ar |
Ar |
Ar |
|
|
O |
O |
|
|
NH |
NH2 |
|
|
Ar |
Ar |
|
SCHEME 10
V. REARRANGEMENT OF N -HALO COMPOUNDS
The Orton rearrangement of N-chloroanilides is well known and the reaction mechanism, at least for reaction in aqueous acid solution, is well understood. The position is set out in Scheme 11. Protonation of the nitrogen atom is followed by nucleophilic attack by the chloride ion, generating chlorine which reacts with the anilide in a conventional electrophilic substitution reaction. The reaction is well documented and the reaction mechanism is well understood58. Only a few recent developments have been reported. A good correlation between log k and has been obtained59 for reactions of 4-X reactants, giving a value of 0.79. This small value is consistent with the conflicting electronic demands of the two stages, the N-protonation and nucleophilic attack by chloride ion. Rearrangement occurs with N-chlorobenzanilide with acid catalysis and chlorine does not migrate to the aromatic ring of the benzyl group60. That part of the reaction leading to de-chlorination has been studied separately61 using triethylamine as the nucleophile and pathways involving the protonated and non-protonated N-chloroacetanilide have been identified.
Rearrangements of N-chloro compounds in heterocyclic systems have been studied.
N-Chloroindole 51 gives62 the |
3-chloro isomer 52. With pyrrole (53) there are two |
|
|
|
Cl |
|
N |
N |
|
|
|
|
Cl |
H |
|
|
|
(51) |
|
(52) |

874 |
D. Lyn H. Williams |
|
|
+ |
|
RCONCl |
RCONHCl |
RCONH |
H + |
Cl |
− |
|
|
+ Cl2 |
X |
X |
X |
RCONH |
RCONH |
|
Cl |
|
+ |
X |
X |
|
Cl |
SCHEME 11
pathways63, one thermal which is believed to be intramolecular giving the 2-chloro compound 54, and an acid-catalysed component leading to the 3-chloro (55) and 2,5-dichloro (56) products. The reaction of N-chlorocarbazole (57) gave a variety of products64, the 3- and 1-chloro compounds and the 3,6- and 1,6-dichlorocarbazoles as well as carbazole itself. Reaction takes place in refluxing methanol and an intermolecular mechanism is argued on the basis of the product distribution. In the presence of added base only the carbazole product of de-chlorination is observed. This is taken to support the idea that the rearrangement reaction is acid-catalysed.
N Cl
Thermal
Intramolecular H
(54)
N
Cl
(53)
Acid Catalysed
Intermolecular
Cl |
|
|
|
+ |
|
|
+ 54 |
N |
Cl |
N |
Cl |
H |
|
H |
|
(55) |
|
(56) |
|

19. Rearrangement reactions involving the amino, nitro and nitroso groups 875
N
Cl
(57)
A quite different mechanism for rearrangement of N-chloro compounds occurs when reaction is carried out in the presence of silver ion. This reaction has been studied by Gassman as part of a quest to identify nitrenium ion intermediates in reactions. The work up to 1970 is covered in a review article65. Rearrangement of the 2-azabicyclo [2.2.1]- heptane derivative 58 occurs readily in a silver ion catalysed process to give the 1-aza derivative 59. Kinetic measurements indicated that heterolytic cleavage had occurred, giving the nitrenium ion and chloride ion. The former then undergoes a skeletal rearrangement typical of these bicyclic systems. Later66 the reaction of N-chloroaniline derivatives were studied, again in MeOH and silver ion assisted. With electron-donating ring substituents the final products are those derived by solvent attack at the ring 2- and 4-positions of the nitrenium ion imminium ion intermediate, but with electron-attracting substituents the corresponding 2- and 4-chloro substitution products are formed. For example, the reaction of 60 gave the 2-chloro isomer 61 (58% yield) together with the de-chlorinated product 62 (26% yield). All of these fit Scheme 12 with the formation of a nitrenium ion followed by nucleophilic attack at the 2- and 4-ring positions. It appears quite unusual that the chloro products are formed in the presence of silver ion, and the authors propose that a tight ionpair (63) is formed from which attack by chloride ion can occur. Kinetic measurements of substituted N-chloroanilines in the thermal reaction67 gave a good correlation of log k with C, giving a large negative value ( 6.35) consistent with the generation of a positive charge on nitrogen, which can be delocalized into the aromatic ring.
|
|
|
A g |
+ |
|
Cl |
|
|
|
|
|
|
|
N |
|
||
|
|
|
MeOH |
|
|
|
||
|
|
|
|
|
|
|
||
|
N |
|
|
|
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
|
(58) |
|
|
|
|
|
(59) |
|
t-Bu |
Cl |
|
t-Bu |
|
|
H |
t-Bu |
H |
|
N |
|
|
|
N |
|
N |
|
Cl
+
CO2 Et |
CO2 Et |
CO2 Et |
(60) |
(61) |
(62) |

876 |
|
D. Lyn H. Williams |
|
R Cl |
R + |
R |
R |
N |
N |
N |
N |
|
|
+ |
|
X |
X |
X |
X + |
|
|
MeOH |
|
|
|
or Cl− |
|
Products
SCHEME 12
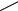 NR
NR
+Cl− Ag+
(63)
VI. REARRANGEMENT INVOLVING NITRO GROUPS
A. The Nitramine Rearrangement
The acid-catalysed rearrangement of N-nitroaniline derivatives continues to provide convenient synthetic routes to some nitro compounds which are difficult to obtain by other methods. A recent example68 is given in Scheme 13, where the introduction of the third nitro group into the aromatic ring is brought about by rearrangement of the
Cl |
|
|
HNCH2 CO2 H |
NO2 |
|
|
NO2 |
NO2 |
+ H2 NCH2 CO2 H |
|
NO2 |
|
|||
|
|
HNO3 /H2 SO4
|
HNCH2 CO2 H |
|
O2 NNCH2 CO2 H |
O2 N |
NO2 |
|
NO2 |
|
|
||
|
|
H2 SO4 |
|
|
NO2 |
|
NO2 |
SCHEME 13