

|
21. Displacement and ipso-substitution in nitration |
959 |
|||
Me |
NO2 |
|
Me |
Me |
|
|
|
|
|
||
|
−NO2 |
− |
+ |
|
H |
|
|
|
|||
|
|
|
|
|
O |
Me |
OAc |
Me |
OAc |
Me |
+C |
|
|
|
|
O |
|
|
(1) |
|
|
|
Me |
|
|
|
|
|
|
|
Me |
|
|
|
|
Me
+
−H +
OAc Me
H OAc
Me
SCHEME 6
Me OR
|
|
|
|
|
Me |
OH |
Me |
NO2 |
|
|
Me |
ROH - H + |
Me |
|
|
|
|
|
||
|
NO |
− |
+ |
+ |
Me - H+ |
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
Me |
Me |
OH |
|
|
Me |
OH |
OH |
|
|
|
|
|
|
|
|
|
|
|
|
OH - H+ |
|
|
|
|
|
|
|
Me |
OH
Me
SCHEME 7

960 |
|
|
J. P. B. Sandall |
|
|
|
|
Me |
A |
Me |
A |
Me |
OMe |
Me |
OH |
|
|
+ |
|
|
|
+ |
|
Me |
NO2 |
Me |
|
Me |
OH |
Me |
OH |
|
|
|
|
|
|
|
|
|
(14) |
|
|
|
(15) |
|
(16) |
|
|
(A = OAc, OH, OMe) |
|
|
|
|
|
|
|
Me |
|
OH |
|
|
|
|
|
|
|
|
|
|
|
A
H
+ Me
Me
Me NO2
H
AcO
Cl
(20)
E
Me NO2
Me
CN
H OAc A A l1
(19)
OH |
Me |
Me |
OH |
|
+ |
Me |
Me |
(17) |
(18) |
SCHEME 8
|
Me |
Me |
|
Me |
Me |
|
+ |
|
|
CN |
CN |
H |
OAc |
OH |
|
|
|
|
(21) |
|
Me
O2 N Me
Me |
Me |
+
CN |
CN |
(22)
SCHEME 9

21. Displacement and ipso-substitution in nitration |
961 |
It is clear that these solvolytic reactions of ipso-adducts are markedly dependent on reaction conditions: strongly acidic conditions and electron withdrawing substituents will favour a heterolytic process; lower acidity, higher temperatures and electron releasing substituents probably favour homolytic processes. With so many factors involved, each substrate under each set of conditions gives rise to its own particular behaviour.
B. Further Addition to the ipso-Species
This aspect of the behaviour of ipso-species has been extensively investigated by Hartshorn’s group. Again, it is impossible to review in any detail all the substrates that have been examined; a few examples will however illustrate the principle processes observed. A typical substrate, 4,5-dichloro-2-methylphenol24, on treatment with concentrated nitric acid gives 2,2,5-trinitro-3,4-dichloro-6-methylcyclohex-3-enone (23). An exactly analogous compound was observed and completely characterized by X-ray crystallography from the nitration of 4,6-dibromo-2,5-dimethylphenol. The IR, and 1H and 13C NMR spectra were fully in accord with this structure; the relative stereochemistry at C5 and C6 was assumed by analogy with that known for the dibromo analogue. Nitration of 4,5-dichloro-2-methyl-6-nitrophenol gave the identical compound. Further evidence for the structure comes from the behaviour of this adduct on heating in tetrachloroethylene:
|
Cl |
|
Cl |
|
CN |
Cl |
NO2 |
Cl |
O |
NC |
Me |
H |
NO2 |
H |
|
Me |
Me |
|
|
||||
|
|
|
|||
O2 N |
O |
O2 N |
O |
O2 N |
O |
|
|
Me |
NO2 |
||
Me |
OH |
Me |
OH |
||
|
(23) |
|
(24) |
|
(25) |
|
|
|
|
O |
|
|
CN |
|
CN |
|
O |
|
|
|
|
||
NC |
Me |
NC |
CH2 ONO2 |
NC |
CH2 |
|
|
||||
Me |
Me |
|
|
|
|
|
|
|
|
|
|
O2 N |
O |
Me |
Me |
Me |
Me |
Me |
OH |
|
Me |
|
Me |
|
|
|
|
||
|
(26) |
|
(27) |
|
(28) |
|
|
CN |
CN |
|
|
|
CN |
Me |
CN |
NO2 |
|
|
|
|
|||
|
|
NO2 |
|
Me |
|
|
Me |
|
Me |
|
|
|
O2 N |
O |
O2 N |
O |
|
|
|
|
|
||
|
Me |
Me |
Me Me |
|
|
|
|
(29) |
(30) |
|
|

962 |
J. P. B. Sandall |
the ˛-diketone (24) is formed with evolution of nitrogen dioxide. This type of addition reaction to the first generated ipso-intermediate is quite general and not restricted to phenols: another example may be found in the nitration of 3,4,5,6-tetramethylbenzene- 1,2-dicarbonitrile. This substrate was first investigated by Suzuki’s group51 who reported the formation of several compounds (25, 26, 27 and 28) on nitration in fuming nitric acid. The structures given for 25 and 26 are not analogous to that, for instance, given above (23). This led Hartshorn and coworkers30 to repeat the reaction; they identified two isomeric dinitroketones (29, 30) and the two corresponding isomeric hydroxy nitro ketones, in addition to the compounds 37 and 38. It will be noted that in this case, all the products have undergone a methyl group migration; the tendency of an ipso-substituted alkyl group to undergo a displacement will be discussed in the next section. The elimination of nitrogen dioxide from the trans dinitro ketone by refluxing in petroleum ether gives the cyclohexa-2,4-dienone (31); regeneration of the cyclohex-3-enones by treatment with nitrogen dioxide gives an approximately equimolar mixture of the cis and trans isomers, 29 and 30. Likewise, the cis dinitroketone rearranges to the trans at 20 °C in chloroform. These rearrangements, and the formation of the hydroxy ketones, were considered to take place via a radical pair intermediate (Scheme 10), the hydroxyl groups arising from hydrolysis of the nitrito ketones.
|
|
CN |
|
CN |
|
|
NO2 |
|
NO |
|
NC |
Me |
NC |
2 |
|
|
|||
29 |
|
|
|
Me |
Me |
|
Me |
|
|
|
|
|
||
|
O2 N |
O |
O2 N |
O |
|
|
|
||
|
Me |
Me |
Me |
Me |
CN |
|
|
CN |
|
NC |
Me |
NC |
ONO |
|
|
|
|||
|
|
|
Me |
|
|
|
Me |
|
|
Me |
O |
O2 N |
O |
|
|
|
|
|
|
Me |
Me |
Me |
Me |
|
(31) |
|
|
|
|
|
|
SCHEME 10 |
|
|
The effects of a |
quite small |
change in structure of the |
substrate may be |
|
illustrated by the behaviour of the 2,3,5,6-tetramethylbenzonitrile30. Here, on treatment with fuming nitric acid, the 6-nitro compound is formed as expected; two other products are the addition compounds 32 and 33, again the cis and trans isomers. Again both compounds can be generated by direct addition of nitrogen dioxide to the 2-cyano-3,4,6,6-tetramethylcyclohexa-2,5-dienone (34). However, the cis dinitroketone dissolves on heating in chloroform to give solely the trans
dinitroketone: clearly in |
this case |
a |
mechanism |
involving homolytic |
loss |
of |
|
nitrogen dioxide is most |
unlikely |
|
such |
a process |
would surely lead to |
loss |
of |
|
|||||||
some nitrogen dioxide and formation of 34. In order to explain the retention of the NO2 moiety during reflux in chloroform, the authors suggest heterorather than homolytic cleavage of the C4 NO2 bond. Thus the rearrangement in this isomer takes place via

|
21. Displacement and ipso-substitution in nitration |
963 |
|||
|
Me |
|
Me |
|
Me |
O2 N |
CN |
Me |
CN Me |
|
CN |
Me |
|
O2 N |
|
|
|
H |
O |
H |
O |
|
O |
O2 N |
O2 N |
|
|||
|
|
|
|
||
Me |
Me |
Me |
Me |
Me |
Me |
|
(32) |
|
(33) |
(34) |
|
an ion pair formed from a nitronium ion and a resonance-stabilized carbanion. Recombination of the two ions with inversion at C-4 gives the required isomer. This difference in behaviour from compounds 29 and 30 above is marked; the extra stabilization of the carbanion is suggested to arise from the ˇ-keto nitrile structure which is noted for the ability to stabilize a carbanionic centre at the ˛-carbon atom this structural feature is absent in 30. Presumably this outweighs the extra stability which may arise in 30 from the second electron withdrawing cyano group. The reaction was also carried out in the presence of mesitylene; no nitromesitylene was formed, demonstrating that the nitronium ion does not escape from the ion pair.
If a more reactive polyalkylphenol is used as substrate, then nitrogen dioxide becomes a useful reagent for the generation and subsequent addition reactions of the ipso- intermediates. Thus reaction of 2,4,6-trialkylphenols31 with nitrogen dioxide generates first the 4-alkyl-4-nitro-2,5-cyclohexadienone which can rearrange to the corresponding 6-nitro compound. Further reaction of this latter compound is by either 1,2- or 1,4- addition of two moles of nitrogen dioxide to form the isomeric trinitro compounds 35 and 36, the final product depending on the sizes of the alkyl groups at C-2 and C-4. Hydroxyketones are also among the products when 4-chloro-2,3,6-trimethylphenol33 is used. In order to characterize these, a reaction was first carried out with nitric acid in acetic acid which favours formation of these compounds relative to the trinitro adducts; four hydroxy ketones were characterized by X-ray crystallography. When the same substrate was reacted with nitrogen dioxide in benzene, in addition to the hydroxy ketones, four isomeric trinitroketones (37 40) were isolated and characterized. These compounds accounted for some 60% of the products and hydroxy ketones for 12%. Comparison of the behaviour of this substrate with that of 3-chloro-2,4,6-trimethylphenol and 2,3,4,6- tetramethylphenol enabled a comparison of the effect of the 3-substituent to be carried out. Essentially the reaction can be explained in terms of the recombination of the phenoxy and NO2 radicals to form either nitro or nitrito compounds, the latter giving rise to hydroxy ketones, the former to nitroketones (Scheme 11), both these species capable of further addition of NO2. For 3-methyl substituted phenols, the pattern of attack was that arising from formation of an ipso-intermediate at the 6-position; for the 3-chloro- substituted phenol, attack must have been at the 2-position. As well as this change in the regiochemistry of the addition, the chloro substituent promotes the formation of
R2 |
R1 |
2 |
NO2 |
R1 |
|
|
NO2 |
R |
|
|
|
|
|
|
|
||
H |
|
H |
|
|
|
O2 N |
O |
O2 N |
O |
||
NO2 |
NO2 |
||||
R3 |
|
R3 |
|||
|
(35) |
|
|
(36) |
|

964 |
|
J. P. B. Sandall |
|
|
|
Cl |
Me |
Cl |
Me |
Cl |
Me |
|
|
|
|||
|
NO2 |
|
NO2 |
|
NO2 |
Me |
|
Me |
|
Me |
|
|
O |
|
O |
|
O |
O2 N Me NO2 |
|
O2 N Me NO2 |
|
O2 N Me NO2 |
|
(37) |
|
(38) |
|
|
(39) |
|
|
Cl |
Me |
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
|
|
|
Me |
|
|
|
|
|
O2 N Me NO2 |
O |
|
|
|
|
|
|
|
|
|
|
(40) |
|
|
|
|
|
Cl |
|
Me |
NO2• |
|
|
|
|
|
|
|
|
NO2• |
|
|
Products |
|
|
|
|
|
|
Cl |
Me |
Me |
|
O |
|
Me |
NO2 |
|
|
||
|
|
|
|
||
Me |
O• |
|
|
|
|
Me |
|
ΟΝΟ• Cl |
|
Me |
NO2• |
|
|
|
|
|
Products |
|
|
Me |
|
O |
|
|
|
Me |
OH |
|
|
SCHEME 11
hydroxy ketones, either by favouring radical recombination via ONO, or by facilitating the hydrolysis.
IV. REARRANGEMENT OF ipso-SUBSTITUTED GROUPS
The migration of ipso-substituted groups, whether those attacked (e.g. alkyl) or those attacking (such as nitro or acetoxyl), is a fertile ground for investigation. The behaviour of a typical ipso-adduct may be conveniently illustrated by the behaviour of the cyclohexadiene adducts arising from the nitration of 4-ethyltoluene in acetic anhydride5. Here all four possible adducts, the diastereoisomeric pairs of the 4-ethyl-1-methyl-4-nitro- and 1- ethyl-4-methyl-4-nitrocyclohexa-2,5-dienyl acetates, were obtained (41, 42) and separated. The authors formulated the migration steps as occurring via the cyclohexadienyl cation. All possible rearrangement products can be realized under appropriate conditions; that is methyl, ethyl, acetoxyl and nitro groups were all observed to migrate. In strongly acidified methanol, the formation of the nitrocyclohexadienyl cation is favoured; in methanol itself, or, better, aqueous methanol, the acetoxycyclohexadienyl cation is produced. The reaction

21. Displacement and ipso-substitution in nitration |
965 |
|||
Me |
OAc |
Me |
NO2 |
|
Et |
NO2 |
Et |
OAc |
(41)(42)
is further complicated by capture of the methoxy group also being of importance, not to mention migrations into the side-chain. However, Fischer and Henderson were able to establish that, for example in the 4-acetoxy-4-alkylcyclohexadienyl cation, 1,2-migration of acetoxyl is faster than alkyl migration, but 1,2-alkyl migration is faster than that of hydroxyl or methoxyl in the corresponding cations. Nitro group migration appears always to take preference over other possibilities.
Much more detailed mechanistic studies have been carried out on the ipso-intermediates, 4-methyl-4-nitrocyclohexa-2,5-dienones (substituted 2-methyl, 3-methyl and 2-nitro), formed by nitration of the corresponding phenols. Here, only nitro group migration is important, and that only from the 4- to the 2-(6-)position. The first careful mechanistic study of such systems was carried out by Barnes and Myhre52 who examined the behaviour on rearrangement of the 4-methyl-, 3,4-dimethyl- and 3,4,5-trimethylcyclohexa- 2,5-dienones in both non-polar organic, aqueous and aqueous-acid solvents. In contrast to the 4-alkyl-4-nitro adducts discussed above, a 1,3-shift of the nitro group rather than a 1,2- shift was observed in all solvents. The reaction was faster in non-polar organic solvents than in water and kinetically first order in ipso-intermediate: the rate of formation of the product 2-nitro compound was equal to the rate of disappearance of the intermediate. The activation entropy was small and positive and radical scavengers lowered the yield of nitro product. Methyl groups substituted in the 3- and 5-positions slowed the reaction down. 15N labelling of the nitro group demonstrated substantial but incomplete scrambling of the label. All these facts were held to be consistent with a radical dissociation recombination process, with extensive loss of NO2 from the solvent cage. An acid-catalysed route for the rearrangement was also found.
The work of Coombes and coworkers20 on the formation of the 4-methyl-4-nitro intermediate has already been discussed above. Here the solvent was aqueous sulphuric acid with acid concentration ranging from 55% to 90%. The final product, 4-methyl-2- nitrophenol, was formed by the expected two routes: about 40% via the ipso-intermediate and 60% directly. Their kinetic studies enabled the acidity dependence of the ipso- rearrangement to be examined; they argued that this dependence demonstrated that the rate-limiting stage of the conversion involved the protonated ipso-intermediate (43). They
Me NO2
+
OH
(43)
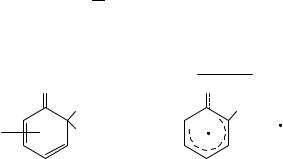
966 |
J. P. B. Sandall |
argued that, unlike the reaction in less acidic media, already established as a radical process, the catalysed process involved a rate-limiting reversion to the nitronium ion/phenol encounter pair, followed by heterolytic fission of the C N bond and fast formation of product. They pointed out that if this acid catalysed process were homolytic, then effectively one must assume that electron transfer from phenol to nitronium ion must take place during encounter pair formation, an earlier suggestion by Perrin53. Later, some evidence that this acid catalysed process did not involve dissociation to the nitronium ion was produced by Myhre54.
In an attempt to clarify these processes further55, the rearrangement of the 4-methyl-4- nitrocyclohexa-2,5-dienone was followed in situ in an NMR spectrometer, to establish the presence of CIDNP effects. The formation of this ipso species has already been discussed; under the conditions of these experiments it is formed essentially by the nitrous acid catalysed radical nitration process, to the extent of approximately 70%, the major side-product being the 4-methyl-2-nitrophenyl acetate (the phenolic product of the rearrangement is acetylated rapidly in the acetic anhydride solvent). The nitrocyclohexadienone also reverts to 4-methylphenyl acetate during this period to the extent of 20%. The reactions are shown in Scheme 1. The rearrangement was followed by 1H NMR; it gave first order kinetics and no sign of any 1H nuclear polarization. Both the uncatalysed and acid-catalysed processes could be distinguished under these conditions; about 4% added sulphuric acid was required to give a reaction proceeding through the catalysed route to the extent of 90%. Substituent effects on this first order rate were also obtained: a 2-methyl substituent increased the rate by 20%, 3-methyl decreased it by about a factor of two and a 2-nitro group increased it by a factor of approximately 20. There was no marked difference between the substituent effect on the catalysed and uncatalysed processes. 15N NMR spectra were then taken during the rearrangement, again under both acid catalysed and non-acid catalysed conditions. The various enhancement factors for the nitrogen nucleus in both reactant and product could be extracted mathematically for all possible routes illustrated in Scheme 1. This showed unambiguously that both the acid catalysed route and the simple thermal rearrangement both give rise to CIDNP effects and both must take place by homolytic dissociation of the C N bond. This is also in accord with the substituent effects mentioned above. It is also clear from these studies that there is a significant return to the initial cyclohexadienone from the radical pair. Although the results require the acid catalysed rearrangement to have a homolytic component, obviously some contribution from a heterolytic process (Path C, Scheme 1) cannot be ruled out.
As we have noted so frequently before, it is important not to extrapolate too far in attempting to deduce a mechanism even when only small structural changes in substrate are involved. The rearrangement of the nitro group in the ipso-intermediates derived from some 2-methylphenol provides an interesting example56. Here the intermediates, variously substituted 2-methyl-2-nitrocyclohexa-3,5-dienones (44), rearrange rapidly and regiospecifically to the corresponding 6-nitro-2-methylphenols unless the 6-position is
|
O |
|
|
O |
|
NO2 |
|
|
Me |
R |
Me |
R |
|
NO2 |
|
||||
|
R = 3-Me, 4-Me, 5-Me, 6-Me |
|
|
|
|
4-NO2 , 6-NO2 |
|
|
|
|
(44) |
|
(45) |
|

21. Displacement and ipso-substitution in nitration |
967 |
blocked. The same regiospecificity is found in the nitro group rearrangements of substituted 2-nitrophenols in trifluoromethanesulphonic acid57, where the rearrangement occurs via the Wheland intermediate. By analogy with the rearrangement of the 4-methyl-4- nitrocyclohexa-2,5-dienones outlined above, one would expect a homolytic mechanism, but a radical pair process involving NO2 is not normally expected to be regiospecific. Such radical pairs as 45, when generated either by rearrangement of an ipso-intermediate or by nitrous acid catalysed nitration, result in attachment of NO2 at both ortho and para positions as would be expected from the very similar spin densities at these sites. Previous workers investigating these substrates had noted the regiospecificity and suggested two possible explanations: either a homolytic process involving the transient formation of phenyl nitrate20 or a 1,5 sigmatropic shift58. By generating the 15N labelled ipso- intermediate and following the 15N NMR spectrum during its rearrangement, it was shown that no CIDNP effect was observable unless the 6-position was blocked, in which case rearrangement takes place to the 4-position with a CIDNP effect. Both the regiospecific 2 6 rearrangement and the clearly homolytic 2 4 rearrangement undergo acid catalysis to much the same extent. It seems that it is most unlikely that completely different mechanisms apply to these two rearrangements: in each case then the first step must involve homolysis of the C N bond. Extensive semi-empirical calculations failed to detect a transition state for a 1,5 sigmatropic process; the NO2 group always appears to move away from the 6-position as the C N bond is stretched. The explanation for the absence of a CIDNP effect, and the marked regiospecificity for the 2 6 rearrangement, must presumably be sought elsewhere. The calculations mentioned above suggested that the barrier to movement of the NO2 towards the 4-position is greater than that for movement towards the oxygen atom of the phenoxy radical. Perhaps then no escape from the radical pair undergoing the 2 6 shift takes place because of stabilizing interactions between the phenoxy oxygen atom and the NO2 nitrogen atom as previously suggested by Coombes and collaborators20. It is worth emphasizing that although the presence of a CIDNP effect can be diagnostic of the presence of a radical pair on the reaction path59, the absence of such effects does not preclude a homolytic reaction. It is also necessary for some escape from the radical pair to take place, for there to be a reasonable electron spin density on the nucleus under observation and for the relaxation time of that nucleus to be sufficiently long. It is for these reasons that the observation of 15N CIDNP effects in nitration processes involving NO2 is easier than with other nuclei in the system.
If the ipso-position attacked during nitration is already substituted by a nitro group, then the consequences of such a process will not be recognized unless the attacking nitrating agent or the original group is labelled (most conveniently with 15N). Thus ipso-attack at the 4-position in 4-nitrophenol was investigated60 and shown to occur to the extent of 20%. The conditions used involved nitric acid in trifluoroacetic acid in the presence of nitrous acid catalysed nitration, so it is not unexpected that the formation of the ipso-intermediate should be a free radical process. The rearrangement of the nitro group to the 2-position was also shown to be a homolytic process, involving the NO2 4-nitrophenoxy radical pair. This was demonstrated by 15N labelling of the 4-nitrophenol and treating this with unlabelled nitric acid; the migrating nitro group in the 2-position showed a CIDNP effect of the appropriate phase. Another interesting aspect of this reaction, although not directly relevant to ipso-attack, is that the 2,4-dinitrophenol which is produced directly, even in conditions when nitrous acid catalysed nitration is prevented, also forms by a homolytic process. The authors suggest that this is possibly via nitronium attack on the phenolic oxygen atom to generate phenyl nitrate, which can then undergo a rearrangement akin to the nitramine rearrangement which is typically a homolytic process. It seems unlikely that this observation of a CIDNP effect arises from direct electron transfer from phenol to nitronium ion, since no such effect is observed with mesitylene, which is expected to be just as easily oxidized.

968 |
J. P. B. Sandall |
The exchange of nitro groups is not restricted to that at the 4-position relative to the hydroxyl group. Konior and coworkers61 have nitrated both 2,6-dinitrophenol and picric acid with 15N labelled nitric acid in acetic anhydride. The product from both nitrations (picric acid) showed the presence of the label in all three positions of the nitro group; indeed, in the picric acid derived from the dinitrophenol, the nitro group in the 4-position was also labelled 14N. No precautions were taken to eliminate nitrous acid catalysed nitration so that it is possible that both formation of the ipso-intermediates and their rearrangement proceed via a radical pair. However, as pointed out by Moodie62, increasing the number of electron withdrawing substituents in the ring may well increase the extent of heterolysis as opposed to homolysis during the apparently very similar nitramine rearrangement; for example, significant heterolysis occurs during the rearrangement of N-2,4-trinitroaniline to picramide.
Yet another way in which an ipso nitro group may rearrange is to generate a nitrito compound. Thus when 2,3,4,5-tetrabromo-6-methylphenol is nitrated in fuming nitric acid, Hartshorn’s group26 noted the formation of the product 2,3,4,5-tetrabromo-6- methyl-6-hydroxycyclohexa-2,4-dienone (46). They suggested that this is produced via the corresponding 6-nitrito intermediate, which could not be isolated since it is hydrolysed during the work-up of the reaction products. Such hydroxy compounds are frequently observed on nitration of alkyl phenols. Confirmation of the existence of these nitrito ipso- intermediates and their facile hydrolysis to the corresponding hydroxy compounds has been gained from analysis of the 1H and 15N NMR spectra obtained63 during the nitrous acid catalysed nitration of 2,6-dichloro-4-methylphenol. Four products were characterized by NMR: 47 50. The 4-nitro-4-methyl ipso-intermediate could be isolated from the nitration; when dissolved in wet chloroform two other compounds are formed in the first five minutes with the disappearance of about half the starting material. One of these is the hydroxy compound 48, the other is clearly an ipso-intermediate. When 15N labelled starting material is used, only two signals are observed; one for the starting material and one at very low field (υ 600 p.p.m.) typical of nitrites. Further evidence for the nature of this compound and for its equilibrium with the nitro intermediate was obtained by treating the hydroxy compound with nitrous acid dissolved in chloroform and again
O |
|
|
O |
|
|
O |
HO |
Br |
Cl |
|
Cl |
Cl |
Cl |
Me |
|
|
|
|
|
|
Br |
Br |
|
|
|
|
|
Br |
|
Me |
NO2 |
|
Me |
OH |
|
|
|
|
|
|
|
(46) |
|
|
(47) |
|
|
(48) |
|
|
O |
|
|
OH |
|
Cl |
|
Cl |
Cl |
|
Cl |
|
Me |
ONO |
CH2 NO2 |
|
|
|
(49) |
|
(50) |